{"title":"GITR exacerbates lysophosphatidylcholine-induced macrophage pyroptosis in sepsis via posttranslational regulation of NLRP3","authors":"Siping Liang, Jinyu Zhou, Can Cao, Yiting Liu, Siqi Ming, Xi Liu, Yuqi Shang, Juanfeng Lao, Qin Peng, Jiahui Yang, Minhao Wu","doi":"10.1038/s41423-024-01170-w","DOIUrl":null,"url":null,"abstract":"The NLRP3 inflammasome functions as an inflammatory driver, but its relationship with lipid metabolic changes in early sepsis remains unclear. Here, we found that GITR expression in monocytes/macrophages was induced by lysophosphatidylcholine (LPC) and was positively correlated with the severity of sepsis. GITR is a costimulatory molecule that is mainly expressed on T cells, but its function in macrophages is largely unknown. Our in vitro data showed that GITR enhanced LPC uptake by macrophages and specifically enhanced NLRP3 inflammasome-mediated macrophage pyroptosis. Furthermore, in vivo studies using either cecal ligation and puncture (CLP) or LPS-induced sepsis models demonstrated that LPC exacerbated sepsis severity/lethality, while conditional knockout of GITR in myeloid cells or NLRP3/caspase-1/IL-1β deficiency attenuated sepsis severity/lethality. Mechanistically, GITR specifically enhanced inflammasome activation by regulating the posttranslational modification (PTM) of NLRP3. GITR competes with NLRP3 for binding to the E3 ligase MARCH7 and recruits MARCH7 to induce deacetylase SIRT2 degradation, leading to decreasing ubiquitination but increasing acetylation of NLRP3. Overall, these findings revealed a novel role of macrophage-derived GITR in regulating the PTM of NLRP3 and systemic inflammatory injury, suggesting that GITR may be a potential therapeutic target for sepsis and other inflammatory diseases.","PeriodicalId":9950,"journal":{"name":"Cellular &Molecular Immunology","volume":"21 7","pages":"674-688"},"PeriodicalIF":19.8000,"publicationDate":"2024-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular &Molecular Immunology","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41423-024-01170-w","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
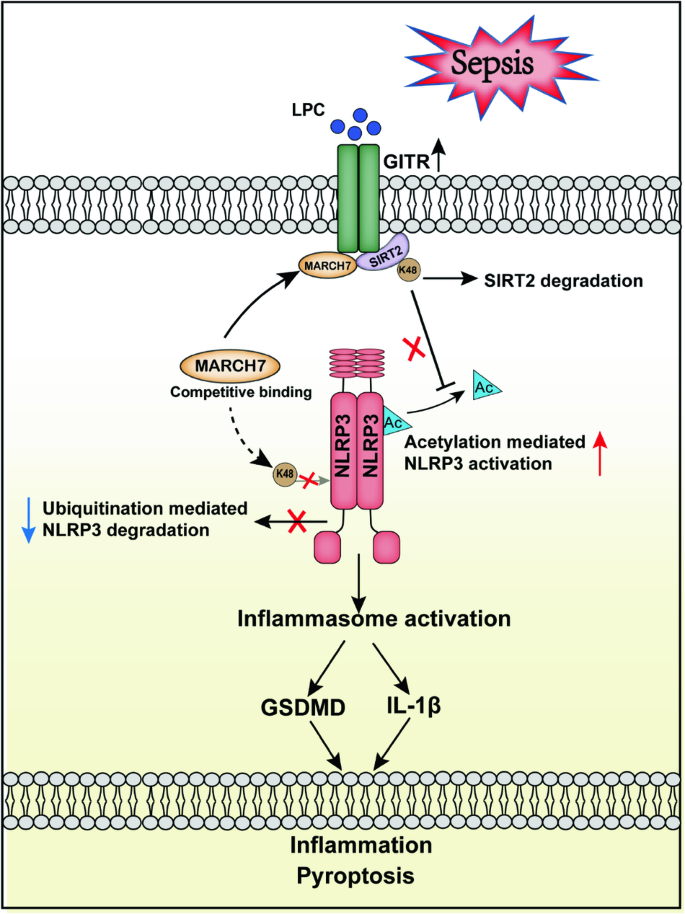
The NLRP3 inflammasome functions as an inflammatory driver, but its relationship with lipid metabolic changes in early sepsis remains unclear. Here, we found that GITR expression in monocytes/macrophages was induced by lysophosphatidylcholine (LPC) and was positively correlated with the severity of sepsis. GITR is a costimulatory molecule that is mainly expressed on T cells, but its function in macrophages is largely unknown. Our in vitro data showed that GITR enhanced LPC uptake by macrophages and specifically enhanced NLRP3 inflammasome-mediated macrophage pyroptosis. Furthermore, in vivo studies using either cecal ligation and puncture (CLP) or LPS-induced sepsis models demonstrated that LPC exacerbated sepsis severity/lethality, while conditional knockout of GITR in myeloid cells or NLRP3/caspase-1/IL-1β deficiency attenuated sepsis severity/lethality. Mechanistically, GITR specifically enhanced inflammasome activation by regulating the posttranslational modification (PTM) of NLRP3. GITR competes with NLRP3 for binding to the E3 ligase MARCH7 and recruits MARCH7 to induce deacetylase SIRT2 degradation, leading to decreasing ubiquitination but increasing acetylation of NLRP3. Overall, these findings revealed a novel role of macrophage-derived GITR in regulating the PTM of NLRP3 and systemic inflammatory injury, suggesting that GITR may be a potential therapeutic target for sepsis and other inflammatory diseases.
期刊介绍:
Cellular & Molecular Immunology, a monthly journal from the Chinese Society of Immunology and the University of Science and Technology of China, serves as a comprehensive platform covering both basic immunology research and clinical applications. The journal publishes a variety of article types, including Articles, Review Articles, Mini Reviews, and Short Communications, focusing on diverse aspects of cellular and molecular immunology.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
