{"title":"A resampling-based approach to share reference panels","authors":"Théo Cavinato, Simone Rubinacci, Anna-Sapfo Malaspinas, Olivier Delaneau","doi":"10.1038/s43588-024-00630-7","DOIUrl":null,"url":null,"abstract":"For many genome-wide association studies, imputing genotypes from a haplotype reference panel is a necessary step. Over the past 15 years, reference panels have become larger and more diverse, leading to improvements in imputation accuracy. However, the latest generation of reference panels is subject to restrictions on data sharing due to concerns about privacy, limiting their usefulness for genotype imputation. In this context, here we propose RESHAPE, a method that employs a recombination Poisson process on a reference panel to simulate the genomes of hypothetical descendants after multiple generations. This data transformation helps to protect against re-identification threats and preserves data attributes, such as linkage disequilibrium patterns and, to some degree, identity-by-descent sharing, allowing for genotype imputation. Our experiments on gold-standard datasets show that simulated descendants up to eight generations can serve as reference panels without substantially reducing genotype imputation accuracy. The authors develop the tool RESHAPE to share reference panels in a safer way. The genome–phenome links in reference panels can generate re-identification threats and RESHAPE breaks these links by shuffling haplotypes while preserving imputation accuracy.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"4 5","pages":"360-366"},"PeriodicalIF":18.3000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s43588-024-00630-7.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-024-00630-7","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
引用次数: 0
Abstract
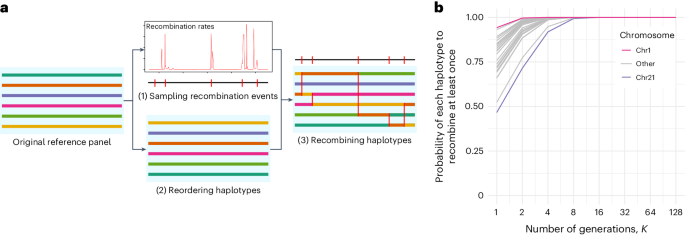
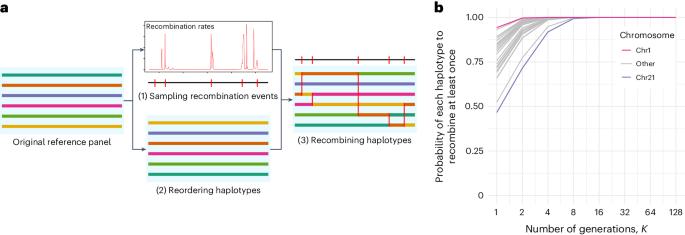
For many genome-wide association studies, imputing genotypes from a haplotype reference panel is a necessary step. Over the past 15 years, reference panels have become larger and more diverse, leading to improvements in imputation accuracy. However, the latest generation of reference panels is subject to restrictions on data sharing due to concerns about privacy, limiting their usefulness for genotype imputation. In this context, here we propose RESHAPE, a method that employs a recombination Poisson process on a reference panel to simulate the genomes of hypothetical descendants after multiple generations. This data transformation helps to protect against re-identification threats and preserves data attributes, such as linkage disequilibrium patterns and, to some degree, identity-by-descent sharing, allowing for genotype imputation. Our experiments on gold-standard datasets show that simulated descendants up to eight generations can serve as reference panels without substantially reducing genotype imputation accuracy. The authors develop the tool RESHAPE to share reference panels in a safer way. The genome–phenome links in reference panels can generate re-identification threats and RESHAPE breaks these links by shuffling haplotypes while preserving imputation accuracy.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
