{"title":"AE-RW: Predicting miRNA-disease associations by using autoencoder and random walk on miRNA-gene-disease heterogeneous network","authors":"Pengli Lu, Jicheng Jiang","doi":"10.1016/j.compbiolchem.2024.108085","DOIUrl":null,"url":null,"abstract":"<div><p>Since scientific investigations have demonstrated that aberrant expression of miRNAs brings about the incidence of numerous intricate diseases, precise determination of miRNA-disease relationships greatly contributes to the advancement of human medical progress. To tackle the issue of inefficient conventional experimental approaches, numerous computational methods have been proposed to predict miRNA-disease association with enhanced accuracy. However, constructing miRNA-gene-disease heterogeneous network by incorporating gene information has been relatively under-explored in existing computational techniques. Accordingly, this paper puts forward a technique to predict miRNA-disease association by applying autoencoder and implementing random walk on miRNA-gene-disease heterogeneous network(AE-RW). Firstly, we integrate association information and similarities between miRNAs, genes, and diseases to construct a miRNA-gene-disease heterogeneous network. Subsequently, we consolidate two network feature representations extracted independently via an autoencoder and a random walk procedure. Finally, deep neural network(DNN) are utilized to conduct association prediction. The experimental results demonstrate that the AE-RW model achieved an AUC of 0.9478 through 5-fold CV on the HMDD v3.2 dataset, outperforming the five most advanced existing models. Additionally, case studies were implemented for breast and lung cancer, further validated the superior predictive capabilities of our model.</p></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"110 ","pages":"Article 108085"},"PeriodicalIF":3.1000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124000732","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
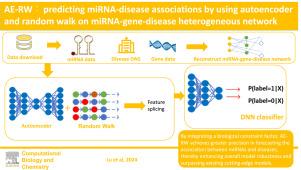
Since scientific investigations have demonstrated that aberrant expression of miRNAs brings about the incidence of numerous intricate diseases, precise determination of miRNA-disease relationships greatly contributes to the advancement of human medical progress. To tackle the issue of inefficient conventional experimental approaches, numerous computational methods have been proposed to predict miRNA-disease association with enhanced accuracy. However, constructing miRNA-gene-disease heterogeneous network by incorporating gene information has been relatively under-explored in existing computational techniques. Accordingly, this paper puts forward a technique to predict miRNA-disease association by applying autoencoder and implementing random walk on miRNA-gene-disease heterogeneous network(AE-RW). Firstly, we integrate association information and similarities between miRNAs, genes, and diseases to construct a miRNA-gene-disease heterogeneous network. Subsequently, we consolidate two network feature representations extracted independently via an autoencoder and a random walk procedure. Finally, deep neural network(DNN) are utilized to conduct association prediction. The experimental results demonstrate that the AE-RW model achieved an AUC of 0.9478 through 5-fold CV on the HMDD v3.2 dataset, outperforming the five most advanced existing models. Additionally, case studies were implemented for breast and lung cancer, further validated the superior predictive capabilities of our model.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
