{"title":"The computational molecular technology for complex reaction systems: The Red Moon approach","authors":"Masataka Nagaoka","doi":"10.1002/wcms.1714","DOIUrl":null,"url":null,"abstract":"<p>For dealing with complex reaction (CR) systems that show typical chemical phenomena in molecular aggregation states, the Red Moon (RM) approach is introduced based on a new efficient and systematic RM methodology. First, the theoretical background with my motivation to develop the RM approach is presented from the recent necessity to perform ‘atomistic’ molecular simulation of large-scale and long-term phenomena of (i) complex chemical reactions, (ii) stereospecificity, and (iii) aggregation structures. The RM methodology uses both the molecular dynamics (MD) method for molecular motions (translation, rotation, and vibration of molecules) that frequently occur on a short-time scale and the Monte Carlo (MC) method for rare events such as chemical reactions that hardly do on that time scale. Then, under the transition rate using both the potential energy difference before and after a rare event trial and its chemical kinetic probability, it is tested and judged by the MC method whether the trial is possible (Metropolis method). Next, typical applications of the RM approach are reviewed in two main research fields, (i) polymerization and (ii) storage battery (rechargeable battery or secondary cell), with various examples of our successful studies. Finally, we conclude that the RM approach using the RM methodology should become an efficient new-generation approach as one promising computational molecular strategy (CMT). We believe it will play an essential role in surveying, at the multilevel resolution, various specificities of CR systems in molecular aggregation states.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 3","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2024-05-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1714","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1714","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
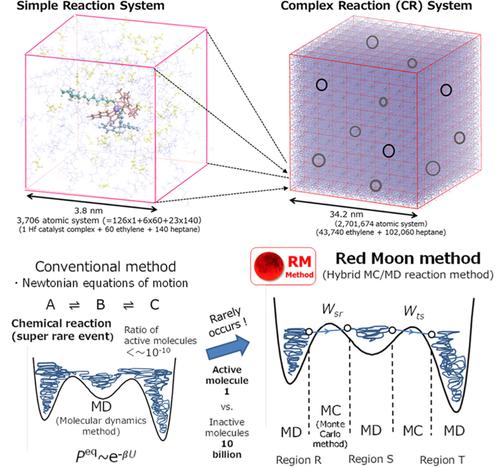
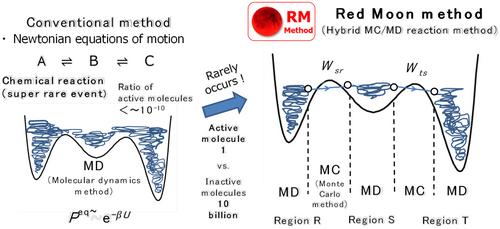
For dealing with complex reaction (CR) systems that show typical chemical phenomena in molecular aggregation states, the Red Moon (RM) approach is introduced based on a new efficient and systematic RM methodology. First, the theoretical background with my motivation to develop the RM approach is presented from the recent necessity to perform ‘atomistic’ molecular simulation of large-scale and long-term phenomena of (i) complex chemical reactions, (ii) stereospecificity, and (iii) aggregation structures. The RM methodology uses both the molecular dynamics (MD) method for molecular motions (translation, rotation, and vibration of molecules) that frequently occur on a short-time scale and the Monte Carlo (MC) method for rare events such as chemical reactions that hardly do on that time scale. Then, under the transition rate using both the potential energy difference before and after a rare event trial and its chemical kinetic probability, it is tested and judged by the MC method whether the trial is possible (Metropolis method). Next, typical applications of the RM approach are reviewed in two main research fields, (i) polymerization and (ii) storage battery (rechargeable battery or secondary cell), with various examples of our successful studies. Finally, we conclude that the RM approach using the RM methodology should become an efficient new-generation approach as one promising computational molecular strategy (CMT). We believe it will play an essential role in surveying, at the multilevel resolution, various specificities of CR systems in molecular aggregation states.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
