Samantha J. Emery-Corbin, Jumana M. Yousef, Subash Adhikari, Fransisca Sumardy, Duong Nhu, Mark F. van Delft, Guillaume Lessene, Jerzy Dziekan, Andrew I. Webb, Laura F. Dagley
{"title":"Improved drug target deconvolution with PISA-DIA using an extended, overlapping temperature gradient","authors":"Samantha J. Emery-Corbin, Jumana M. Yousef, Subash Adhikari, Fransisca Sumardy, Duong Nhu, Mark F. van Delft, Guillaume Lessene, Jerzy Dziekan, Andrew I. Webb, Laura F. Dagley","doi":"10.1002/pmic.202300644","DOIUrl":null,"url":null,"abstract":"<p>Thermal proteome profiling (TPP) is a powerful tool for drug target deconvolution. Recently, data-independent acquisition mass spectrometry (DIA-MS) approaches have demonstrated significant improvements to depth and missingness in proteome data, but traditional TPP (a.k.a. CEllular Thermal Shift Assay “CETSA”) workflows typically employ multiplexing reagents reliant on data-dependent acquisition (DDA). Herein, we introduce a new experimental design for the Proteome Integral Solubility Alteration via label-free DIA approach (PISA-DIA). We highlight the proteome coverage and sensitivity achieved by using multiple overlapping thermal gradients alongside DIA-MS, which maximizes efficiencies in PISA sample concatenation and safeguards against missing protein targets that exist at high melting temperatures. We demonstrate our extended PISA-DIA design has superior proteome coverage as compared to using tandem-mass tags (TMT) necessitating DDA-MS analysis. Importantly, we demonstrate our PISA-DIA approach has the quantitative and statistical rigor using A-1331852, a specific inhibitor of BCL-xL. Due to the high melt temperature of this protein target, we utilized our extended multiple gradient PISA-DIA workflow to identify BCL-xL. We assert our novel overlapping gradient PISA-DIA-MS approach is ideal for unbiased drug target deconvolution, spanning a large temperature range whilst minimizing target dropout between gradients, increasing the likelihood of resolving the protein targets of novel compounds.</p>","PeriodicalId":224,"journal":{"name":"Proteomics","volume":"24 16","pages":""},"PeriodicalIF":3.9000,"publicationDate":"2024-05-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/pmic.202300644","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics","FirstCategoryId":"99","ListUrlMain":"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/pmic.202300644","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
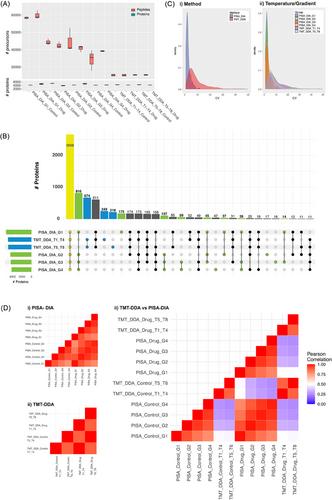
Thermal proteome profiling (TPP) is a powerful tool for drug target deconvolution. Recently, data-independent acquisition mass spectrometry (DIA-MS) approaches have demonstrated significant improvements to depth and missingness in proteome data, but traditional TPP (a.k.a. CEllular Thermal Shift Assay “CETSA”) workflows typically employ multiplexing reagents reliant on data-dependent acquisition (DDA). Herein, we introduce a new experimental design for the Proteome Integral Solubility Alteration via label-free DIA approach (PISA-DIA). We highlight the proteome coverage and sensitivity achieved by using multiple overlapping thermal gradients alongside DIA-MS, which maximizes efficiencies in PISA sample concatenation and safeguards against missing protein targets that exist at high melting temperatures. We demonstrate our extended PISA-DIA design has superior proteome coverage as compared to using tandem-mass tags (TMT) necessitating DDA-MS analysis. Importantly, we demonstrate our PISA-DIA approach has the quantitative and statistical rigor using A-1331852, a specific inhibitor of BCL-xL. Due to the high melt temperature of this protein target, we utilized our extended multiple gradient PISA-DIA workflow to identify BCL-xL. We assert our novel overlapping gradient PISA-DIA-MS approach is ideal for unbiased drug target deconvolution, spanning a large temperature range whilst minimizing target dropout between gradients, increasing the likelihood of resolving the protein targets of novel compounds.
期刊介绍:
PROTEOMICS is the premier international source for information on all aspects of applications and technologies, including software, in proteomics and other "omics". The journal includes but is not limited to proteomics, genomics, transcriptomics, metabolomics and lipidomics, and systems biology approaches. Papers describing novel applications of proteomics and integration of multi-omics data and approaches are especially welcome.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
