Nikolai V. Krivoshchapov*, and , Michael G. Medvedev*,
{"title":"Accurate and Efficient Conformer Sampling of Cyclic Drug-Like Molecules with Inverse Kinematics","authors":"Nikolai V. Krivoshchapov*, and , Michael G. Medvedev*, ","doi":"10.1021/acs.jcim.3c02040","DOIUrl":null,"url":null,"abstract":"<p >Identification of all of the influential conformers of biomolecules is a crucial step in many tasks of computational biochemistry. Specifically, molecular docking, a key component of <i>in silico</i> drug development, requires a comprehensive set of conformations for potential candidates in order to generate the optimal ligand–receptor poses and, ultimately, find the best drug candidates. However, the presence of flexible cycles in a molecule complicates the initial search for conformers since exhaustive sampling algorithms <i>via</i> torsional random and systematic searches become very inefficient. The devised inverse-kinematics-based Monte Carlo with refinement (MCR) algorithm identifies independently rotatable dihedral angles in (poly)cyclic molecules and uses them to perform global conformational sampling, outperforming popular alternatives (MacroModel, CREST, and RDKit) in terms of speed and diversity of the resulting conformer ensembles. Moreover, MCR quickly and accurately recovers naturally occurring macrocycle conformations for most of the considered molecules.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 11","pages":"4542–4552"},"PeriodicalIF":5.3000,"publicationDate":"2024-05-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.3c02040","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
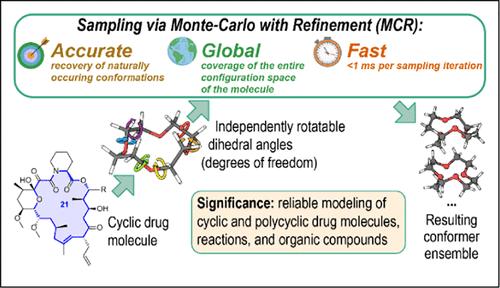
Identification of all of the influential conformers of biomolecules is a crucial step in many tasks of computational biochemistry. Specifically, molecular docking, a key component of in silico drug development, requires a comprehensive set of conformations for potential candidates in order to generate the optimal ligand–receptor poses and, ultimately, find the best drug candidates. However, the presence of flexible cycles in a molecule complicates the initial search for conformers since exhaustive sampling algorithms via torsional random and systematic searches become very inefficient. The devised inverse-kinematics-based Monte Carlo with refinement (MCR) algorithm identifies independently rotatable dihedral angles in (poly)cyclic molecules and uses them to perform global conformational sampling, outperforming popular alternatives (MacroModel, CREST, and RDKit) in terms of speed and diversity of the resulting conformer ensembles. Moreover, MCR quickly and accurately recovers naturally occurring macrocycle conformations for most of the considered molecules.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
