Stefano Bosio, Mattia Bernetti*, Walter Rocchia and Matteo Masetti,
{"title":"Similarities and Differences in Ligand Binding to Protein and RNA Targets: The Case of Riboflavin","authors":"Stefano Bosio, Mattia Bernetti*, Walter Rocchia and Matteo Masetti, ","doi":"10.1021/acs.jcim.4c00420","DOIUrl":null,"url":null,"abstract":"<p >It is nowadays clear that RNA molecules can play active roles in several biological processes. As a result, an increasing number of RNAs are gradually being identified as potentially druggable targets. In particular, noncoding RNAs can adopt highly organized conformations that are suitable for drug binding. However, RNAs are still considered challenging targets due to their complex structural dynamics and high charge density. Thus, elucidating relevant features of drug-RNA binding is fundamental for advancing drug discovery. Here, by using Molecular Dynamics simulations, we compare key features of ligand binding to proteins with those observed in RNA. Specifically, we explore similarities and differences in terms of (i) conformational flexibility of the target, (ii) electrostatic contribution to binding free energy, and (iii) water and ligand dynamics. As a test case, we examine binding of the same ligand, namely riboflavin, to protein and RNA targets, specifically the riboflavin (RF) kinase and flavin mononucleotide (FMN) riboswitch. The FMN riboswitch exhibited enhanced fluctuations and explored a wider conformational space, compared to the protein target, underscoring the importance of RNA flexibility in ligand binding. Conversely, a similar electrostatic contribution to the binding free energy of riboflavin was found. Finally, greater stability of water molecules was observed in the FMN riboswitch compared to the RF kinase, possibly due to the different shape and polarity of the pockets.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 11","pages":"4570–4586"},"PeriodicalIF":5.3000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00420","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
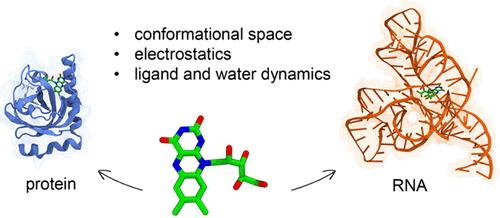
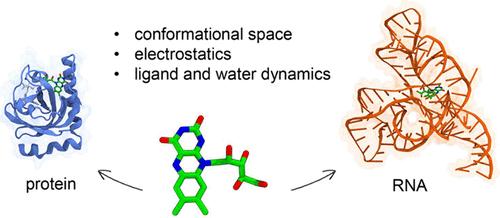
It is nowadays clear that RNA molecules can play active roles in several biological processes. As a result, an increasing number of RNAs are gradually being identified as potentially druggable targets. In particular, noncoding RNAs can adopt highly organized conformations that are suitable for drug binding. However, RNAs are still considered challenging targets due to their complex structural dynamics and high charge density. Thus, elucidating relevant features of drug-RNA binding is fundamental for advancing drug discovery. Here, by using Molecular Dynamics simulations, we compare key features of ligand binding to proteins with those observed in RNA. Specifically, we explore similarities and differences in terms of (i) conformational flexibility of the target, (ii) electrostatic contribution to binding free energy, and (iii) water and ligand dynamics. As a test case, we examine binding of the same ligand, namely riboflavin, to protein and RNA targets, specifically the riboflavin (RF) kinase and flavin mononucleotide (FMN) riboswitch. The FMN riboswitch exhibited enhanced fluctuations and explored a wider conformational space, compared to the protein target, underscoring the importance of RNA flexibility in ligand binding. Conversely, a similar electrostatic contribution to the binding free energy of riboflavin was found. Finally, greater stability of water molecules was observed in the FMN riboswitch compared to the RF kinase, possibly due to the different shape and polarity of the pockets.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
