Kae R. Whiting, Lonneke Haer-Wigman, Ralph J. Florijn, Ronald van Beek, Machteld M. Oud, Astrid S. Plomp, Camiel J. F. Boon, Hester Y. Kroes, Ronald Roepman
{"title":"Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa","authors":"Kae R. Whiting, Lonneke Haer-Wigman, Ralph J. Florijn, Ronald van Beek, Machteld M. Oud, Astrid S. Plomp, Camiel J. F. Boon, Hester Y. Kroes, Ronald Roepman","doi":"10.1038/s41431-024-01627-6","DOIUrl":null,"url":null,"abstract":"INPP5E encodes inositol polyphosphate-5-phosphatase E, an enzyme involved in regulating the phosphatidylinositol (PIP) makeup of the primary cilium membrane. Pathogenic variants in INPP5E hence cause a variety of ciliopathies: genetic disorders caused by dysfunctional cilia. While the majority of these disorders are syndromic, such as the neuronal ciliopathy Joubert syndrome, in some cases patients will present with an isolated phenotype—most commonly non-syndromic retinitis pigmentosa (RP). Here, we report two novel variants in INPP5E identified in two patients with non-syndromic RP: patient 1 with compound heterozygous variants (c.1516C > T, p.(Q506*), and c.847G > A, p.(A283T)) and patient 2 with a homozygous variant (c.1073C > T, p.(P358L)). To determine whether these variants were causative for the phenotype in the patients, automated ciliary phenotyping of patient-derived dermal fibroblasts was performed for percent ciliation, cilium length, retrograde IFT trafficking, and INPP5E localization. In both patients, a decrease in ciliary length and loss of INPP5E localization in the primary cilia were seen. With these molecular findings, we can confirm functionally that the novel variants in INPP5E are causative for the RP phenotypes seen in both patients. Additionally, this study demonstrates the usefulness of utilizing ciliary phenotyping as an assistant in ciliopathy diagnosis and phenotyping.","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"32 11","pages":"1412-1418"},"PeriodicalIF":3.7000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41431-024-01627-6.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41431-024-01627-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
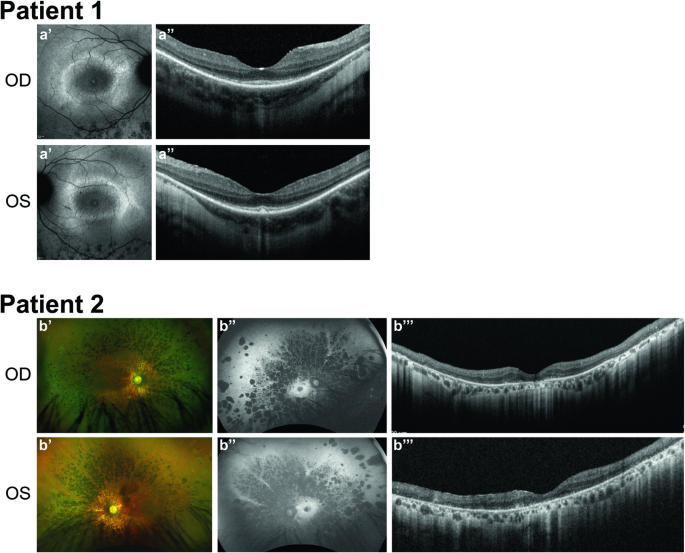
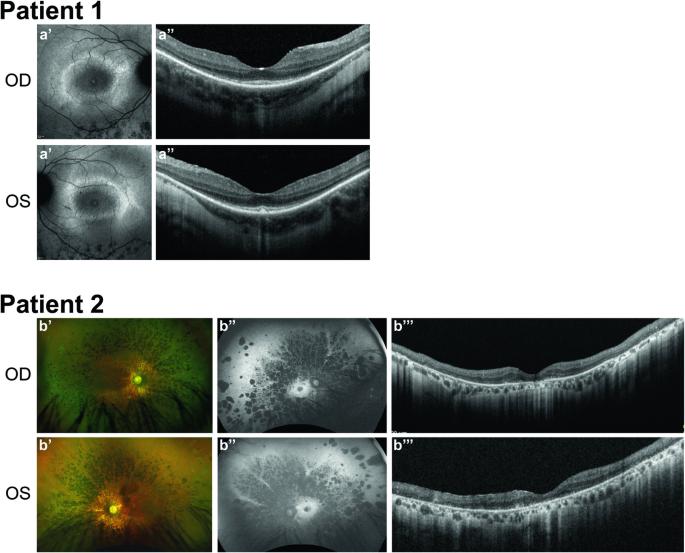
INPP5E encodes inositol polyphosphate-5-phosphatase E, an enzyme involved in regulating the phosphatidylinositol (PIP) makeup of the primary cilium membrane. Pathogenic variants in INPP5E hence cause a variety of ciliopathies: genetic disorders caused by dysfunctional cilia. While the majority of these disorders are syndromic, such as the neuronal ciliopathy Joubert syndrome, in some cases patients will present with an isolated phenotype—most commonly non-syndromic retinitis pigmentosa (RP). Here, we report two novel variants in INPP5E identified in two patients with non-syndromic RP: patient 1 with compound heterozygous variants (c.1516C > T, p.(Q506*), and c.847G > A, p.(A283T)) and patient 2 with a homozygous variant (c.1073C > T, p.(P358L)). To determine whether these variants were causative for the phenotype in the patients, automated ciliary phenotyping of patient-derived dermal fibroblasts was performed for percent ciliation, cilium length, retrograde IFT trafficking, and INPP5E localization. In both patients, a decrease in ciliary length and loss of INPP5E localization in the primary cilia were seen. With these molecular findings, we can confirm functionally that the novel variants in INPP5E are causative for the RP phenotypes seen in both patients. Additionally, this study demonstrates the usefulness of utilizing ciliary phenotyping as an assistant in ciliopathy diagnosis and phenotyping.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
