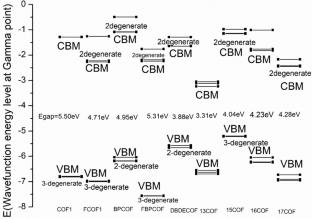
{"title":"Electronic structures of two-dimensional π-conjugated boroxine covalent organic frameworks","authors":"Qi Qi, Li Junfeng, Sun Yueming","doi":"10.1007/s11224-024-02344-y","DOIUrl":null,"url":null,"abstract":"<div><p>Boroxine covalent organic frameworks (boroxine COFs) can be referred to as two-dimension (2D) polymer networks with cores of boroxine connected with rigid linker and extended in x or y dimensions, which might be good candidates for organic electronic and luminescence material. However, the engineering COFs with improved charge transfer and conduction properties based on pore size and linker structure still confront various challenges. Here, we investigate the geometrical structure and electronic properties with linkers from phenyl to biphenyl and the replacement of H with F atom in 2D scale with the CRYTSTAL17 software at the density functional theory (DFT) level with the global-hybrid PBE0 functional and POB-TZVP basis set, using a 5 × 5 k-point mesh. The increased pore diameter reduced their energy gap. Replacing the F atom with the H atom lowered their VBMs and conduction band minimums (CBMs). Computation results can assist the experimental scientist in producing highly conductive 2D boroxine COFs.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 6","pages":"1991 - 2000"},"PeriodicalIF":2.2000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02344-y","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Boroxine covalent organic frameworks (boroxine COFs) can be referred to as two-dimension (2D) polymer networks with cores of boroxine connected with rigid linker and extended in x or y dimensions, which might be good candidates for organic electronic and luminescence material. However, the engineering COFs with improved charge transfer and conduction properties based on pore size and linker structure still confront various challenges. Here, we investigate the geometrical structure and electronic properties with linkers from phenyl to biphenyl and the replacement of H with F atom in 2D scale with the CRYTSTAL17 software at the density functional theory (DFT) level with the global-hybrid PBE0 functional and POB-TZVP basis set, using a 5 × 5 k-point mesh. The increased pore diameter reduced their energy gap. Replacing the F atom with the H atom lowered their VBMs and conduction band minimums (CBMs). Computation results can assist the experimental scientist in producing highly conductive 2D boroxine COFs.
硼氧共价有机框架(Boroxine covalent organic frameworks,简称硼氧框架)可以被称为二维(2D)聚合物网络,其核心是由刚性连接体连接并在 x 或 y 维上延伸的硼氧,可能是有机电子和发光材料的良好候选材料。然而,如何根据孔隙大小和连接体结构设计出具有更好电荷转移和传导性能的 COF,仍然面临着各种挑战。在此,我们利用 CRYTSTAL17 软件,在密度泛函理论(DFT)水平上,使用全局混合 PBE0 函数和 POB-TZVP 基集,使用 5 × 5 k 点网格,研究了从苯基到联苯的连接体以及用 F 原子取代 H 原子的二维尺度几何结构和电子特性。孔径的增大减小了它们的能隙。用 H 原子取代 F 原子降低了它们的 VBM 和导带最小值(CBM)。计算结果有助于实验科学家制备高导电性的二维硼氧 COF。
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
