Tianbai Huang, Robert Geitner, Alexander Croy and Stefanie Gräfe
{"title":"Tailoring phosphine ligands for improved C–H activation: insights from Δ-machine learning†","authors":"Tianbai Huang, Robert Geitner, Alexander Croy and Stefanie Gräfe","doi":"10.1039/D4DD00037D","DOIUrl":null,"url":null,"abstract":"<p >Transition metal complexes have played crucial roles in various homogeneous catalytic processes due to their exceptional versatility. This adaptability stems not only from the central metal ions but also from the vast array of choices of the ligand spheres, which form an enormously large chemical space. For example, Rh complexes, with a well-designed ligand sphere, are known to be efficient in catalyzing the C–H activation process in alkanes. To investigate the structure–property relation of the Rh complex and identify the optimal ligand that minimizes the calculated reaction energy Δ<em>E</em> of an alkane C–H activation, we have applied a Δ-machine learning method trained on various features to study 1743 pairs of reactants (Rh(PLP)(Cl)(CO)) and intermediates (Rh(PLP)(Cl)(CO)(H)(propyl)). Our findings demonstrate that the models exhibit robust predictive performance when trained on features derived from electron density (<em>R</em><small><sup>2</sup></small> = 0.816), and SOAPs (<em>R</em><small><sup>2</sup></small> = 0.819), a set of position-based descriptors. Leveraging the model trained on xTB-SOAPs that only depend on the xTB-equilibrium structures, we propose an efficient and accurate screening procedure to explore the extensive chemical space of bisphosphine ligands. By applying this screening procedure, we identify ten newly selected reactant–intermediate pairs with an average Δ<em>E</em> of 33.2 kJ mol<small><sup>−1</sup></small>, remarkably lower than the average Δ<em>E</em> of the original data set of 68.0 kJ mol<small><sup>−1</sup></small>. This underscores the efficacy of our screening procedure in pinpointing structures with significantly lower energy levels.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 7","pages":" 1350-1364"},"PeriodicalIF":6.2000,"publicationDate":"2024-05-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00037d?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00037d","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
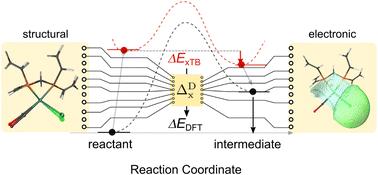
Transition metal complexes have played crucial roles in various homogeneous catalytic processes due to their exceptional versatility. This adaptability stems not only from the central metal ions but also from the vast array of choices of the ligand spheres, which form an enormously large chemical space. For example, Rh complexes, with a well-designed ligand sphere, are known to be efficient in catalyzing the C–H activation process in alkanes. To investigate the structure–property relation of the Rh complex and identify the optimal ligand that minimizes the calculated reaction energy ΔE of an alkane C–H activation, we have applied a Δ-machine learning method trained on various features to study 1743 pairs of reactants (Rh(PLP)(Cl)(CO)) and intermediates (Rh(PLP)(Cl)(CO)(H)(propyl)). Our findings demonstrate that the models exhibit robust predictive performance when trained on features derived from electron density (R2 = 0.816), and SOAPs (R2 = 0.819), a set of position-based descriptors. Leveraging the model trained on xTB-SOAPs that only depend on the xTB-equilibrium structures, we propose an efficient and accurate screening procedure to explore the extensive chemical space of bisphosphine ligands. By applying this screening procedure, we identify ten newly selected reactant–intermediate pairs with an average ΔE of 33.2 kJ mol−1, remarkably lower than the average ΔE of the original data set of 68.0 kJ mol−1. This underscores the efficacy of our screening procedure in pinpointing structures with significantly lower energy levels.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
