Ji-Hee Yoon, Soojin Hwang, Hyunwoo Bae, Dohyung Kim, Go Hun Seo, June-Young Koh, Young Seok Ju, Hyo-Sang Do, Soyoung Kim, Gu-Hwan Kim, Ja Hye Kim, Jin-Ho Choi, Beom Hee Lee
{"title":"Clinical and molecular characteristics of Korean patients with Kabuki syndrome","authors":"Ji-Hee Yoon, Soojin Hwang, Hyunwoo Bae, Dohyung Kim, Go Hun Seo, June-Young Koh, Young Seok Ju, Hyo-Sang Do, Soyoung Kim, Gu-Hwan Kim, Ja Hye Kim, Jin-Ho Choi, Beom Hee Lee","doi":"10.1038/s10038-024-01258-1","DOIUrl":null,"url":null,"abstract":"Kabuki syndrome (KS) is a rare disorder characterized by typical facial features, skeletal anomalies, fetal fingertip pad persistence, postnatal growth retardation, and intellectual disabilities. Heterozygous variants of the KMT2D and KDM6A genes are major genetic causes of KS. This study aimed to report the clinical and genetic characteristics of KS. This study included 28 Korean patients (14 boys and 14 girls) with KS through molecular genetic testing, including direct Sanger sequencing, whole-exome sequencing, or whole-genome sequencing. The median age at clinical diagnosis was 18.5 months (IQR 7–58 months), and the median follow-up duration was 80.5 months (IQR 48–112 months). Molecular genetic testing identified different pathogenic variants of the KMT2D (n = 23) and KDM6A (n = 3) genes, including 15 novel variants. Patients showed typical facial features (100%), such as long palpebral fissure and eversion of the lower eyelid; intellectual disability/developmental delay (96%); short stature (79%); and congenital cardiac anomalies (75%). Although 71% experienced failure to thrive in infancy, 54% of patients showed a tendency toward overweight/obesity in early childhood. Patients with KDM6A variants demonstrated severe genotype-phenotype correlation. This study enhances the understanding of the clinical and genetic characteristics of KS.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 9","pages":"417-423"},"PeriodicalIF":2.5000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01258-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
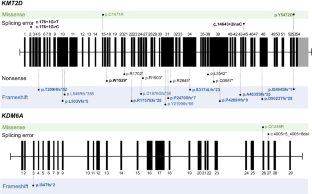
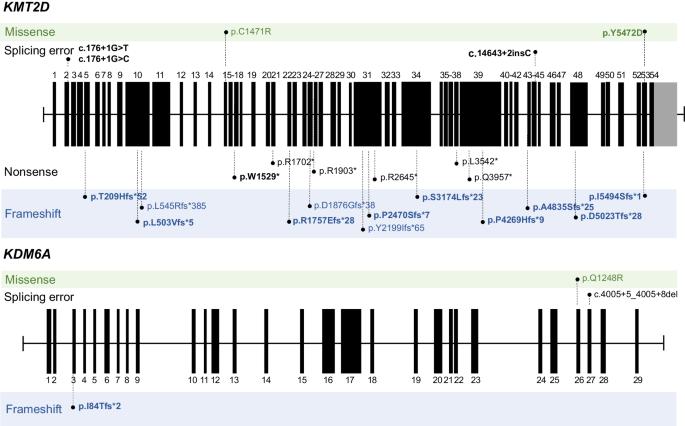
Kabuki syndrome (KS) is a rare disorder characterized by typical facial features, skeletal anomalies, fetal fingertip pad persistence, postnatal growth retardation, and intellectual disabilities. Heterozygous variants of the KMT2D and KDM6A genes are major genetic causes of KS. This study aimed to report the clinical and genetic characteristics of KS. This study included 28 Korean patients (14 boys and 14 girls) with KS through molecular genetic testing, including direct Sanger sequencing, whole-exome sequencing, or whole-genome sequencing. The median age at clinical diagnosis was 18.5 months (IQR 7–58 months), and the median follow-up duration was 80.5 months (IQR 48–112 months). Molecular genetic testing identified different pathogenic variants of the KMT2D (n = 23) and KDM6A (n = 3) genes, including 15 novel variants. Patients showed typical facial features (100%), such as long palpebral fissure and eversion of the lower eyelid; intellectual disability/developmental delay (96%); short stature (79%); and congenital cardiac anomalies (75%). Although 71% experienced failure to thrive in infancy, 54% of patients showed a tendency toward overweight/obesity in early childhood. Patients with KDM6A variants demonstrated severe genotype-phenotype correlation. This study enhances the understanding of the clinical and genetic characteristics of KS.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
