Andrea Gazzin, Federico Fornari, Marcello Niceta, Chiara Leoni, Maria Lisa Dentici, Diana Carli, Anna Maria Villar, Giulio Calcagni, Elena Banaudi, Stefania Massuras, Simona Cardaropoli, Elena Airulo, Paola Daniele, Emanuele Monda, Giuseppe Limongelli, Chiara Riggi, Giuseppe Zampino, Maria Cristina Digilio, Alessandro De Luca, Marco Tartaglia, Giovanni Battista Ferrero, Alessandro Mussa
{"title":"Defining the variant-phenotype correlation in patients affected by Noonan syndrome with the RAF1:c.770C>T p.(Ser257Leu) variant","authors":"Andrea Gazzin, Federico Fornari, Marcello Niceta, Chiara Leoni, Maria Lisa Dentici, Diana Carli, Anna Maria Villar, Giulio Calcagni, Elena Banaudi, Stefania Massuras, Simona Cardaropoli, Elena Airulo, Paola Daniele, Emanuele Monda, Giuseppe Limongelli, Chiara Riggi, Giuseppe Zampino, Maria Cristina Digilio, Alessandro De Luca, Marco Tartaglia, Giovanni Battista Ferrero, Alessandro Mussa","doi":"10.1038/s41431-024-01643-6","DOIUrl":null,"url":null,"abstract":"Hypertrophic cardiomyopathy (HCM) is the major contributor to morbidity and mortality in Noonan syndrome (NS). Gain-of-function variants in RAF1 are associated with high prevalence of HCM. Among these, NM_002880.4:c.770C > T, NP_002871.1:p.(Ser257Leu) accounts for approximately half of cases and has been reported as associated with a particularly severe outcome. Nevertheless, comprehensive studies on cases harboring this variant are missing. To precisely define the phenotype associated to the RAF1:c.770C > T, variant, an observational retrospective analysis on patients carrying the c.770C > T variant was conducted merging 17 unpublished patients and literature-derived ones. Data regarding prenatal findings, clinical features and cardiac phenotypes were collected to provide an exhaustive description of the associated phenotype. Clinical information was collected in 107 patients. Among them, 92% had HCM, mostly diagnosed within the first year of life. Thirty percent of patients were preterm and 47% of the newborns was admitted in a neonatal intensive care unit, mainly due to respiratory complications of HCM and/or pulmonary arterial hypertension. Mortality rate was 13%, mainly secondary to HCM-related complications (62%) at the average age of 7.5 months. Short stature had a prevalence of 91%, while seizures and ID of 6% and 12%, respectively. Two cases out of 75 (3%) developed neoplasms. In conclusion, patients with the RAF1:c.770C > T pathogenic variant show a particularly severe phenotype characterized by rapidly progressive neonatal HCM and high mortality rate suggesting the necessity of careful monitoring and early intervention to prevent or slow down the progression of HCM.","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"32 8","pages":"964-971"},"PeriodicalIF":3.7000,"publicationDate":"2024-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41431-024-01643-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
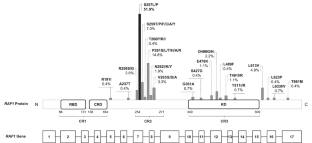
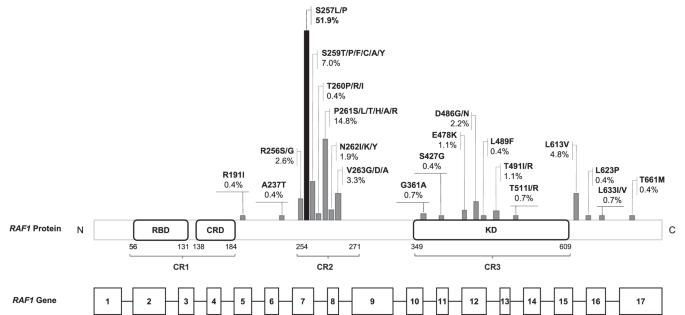
Hypertrophic cardiomyopathy (HCM) is the major contributor to morbidity and mortality in Noonan syndrome (NS). Gain-of-function variants in RAF1 are associated with high prevalence of HCM. Among these, NM_002880.4:c.770C > T, NP_002871.1:p.(Ser257Leu) accounts for approximately half of cases and has been reported as associated with a particularly severe outcome. Nevertheless, comprehensive studies on cases harboring this variant are missing. To precisely define the phenotype associated to the RAF1:c.770C > T, variant, an observational retrospective analysis on patients carrying the c.770C > T variant was conducted merging 17 unpublished patients and literature-derived ones. Data regarding prenatal findings, clinical features and cardiac phenotypes were collected to provide an exhaustive description of the associated phenotype. Clinical information was collected in 107 patients. Among them, 92% had HCM, mostly diagnosed within the first year of life. Thirty percent of patients were preterm and 47% of the newborns was admitted in a neonatal intensive care unit, mainly due to respiratory complications of HCM and/or pulmonary arterial hypertension. Mortality rate was 13%, mainly secondary to HCM-related complications (62%) at the average age of 7.5 months. Short stature had a prevalence of 91%, while seizures and ID of 6% and 12%, respectively. Two cases out of 75 (3%) developed neoplasms. In conclusion, patients with the RAF1:c.770C > T pathogenic variant show a particularly severe phenotype characterized by rapidly progressive neonatal HCM and high mortality rate suggesting the necessity of careful monitoring and early intervention to prevent or slow down the progression of HCM.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
