{"title":"Peptide Drug Design Using Alchemical Free Energy Calculation: An Application and Validation on Agonists of Ghrelin Receptor","authors":"Qin Zeng, Guangpeng Meng, Bingyu Zhao, Haodian Lin, Yuqing Guan, Xiaobin Qin, Yu Yuan*, Yuanbo Li* and Qiantao Wang*, ","doi":"10.1021/acs.jcim.4c00414","DOIUrl":null,"url":null,"abstract":"<p >With recent large-scale applications and validations, the relative binding free energy (RBFE) calculated using alchemical free energy methods has been proven to be an accurate measure to probe the binding of small-molecule drug candidates. On the other hand, given the flexibility of peptides, it is of great interest to find out whether sufficient sampling could be achieved within the typical time scale of such calculation, and a similar level of accuracy could be reached for peptide drugs. However, the systematic evaluation of such calculations on protein–peptide systems has been less reported. Most reported studies of peptides were restricted to a limited number of data points or lacking experimental support. To demonstrate the applicability of the alchemical free energy method for protein–peptide systems in a typical real-world drug discovery project, we report an application of the thermodynamic integration (TI) method to the RBFE calculation of ghrelin receptor and its peptide agonists. Along with the calculation, the synthesis and in vitro EC<sub>50</sub> activity of relamorelin and 17 new peptide derivatives were also reported. A cost-effective criterion to determine the data collection time was proposed for peptides in the TI simulation. The average of three TI repeats yielded a mean absolute error of 0.98 kcal/mol and Pearson’s correlation coefficient (<i>R</i>) of 0.77 against the experimental free energy derived from the in vitro EC<sub>50</sub> activity, showing good repeatability of the proposed method and a slightly better agreement than the results obtained from the arbitrary time frames up to 20 ns. Although it is limited by having one target and a deduced binding pose, we hope that this study can add some insights into alchemical free energy calculation of protein–peptide systems, providing theoretical assistance to the development of peptide drugs.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 12","pages":"4863–4876"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00414","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
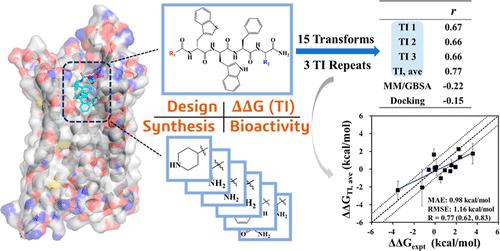
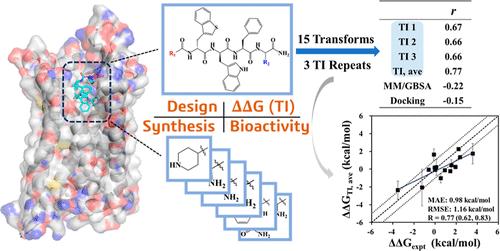
With recent large-scale applications and validations, the relative binding free energy (RBFE) calculated using alchemical free energy methods has been proven to be an accurate measure to probe the binding of small-molecule drug candidates. On the other hand, given the flexibility of peptides, it is of great interest to find out whether sufficient sampling could be achieved within the typical time scale of such calculation, and a similar level of accuracy could be reached for peptide drugs. However, the systematic evaluation of such calculations on protein–peptide systems has been less reported. Most reported studies of peptides were restricted to a limited number of data points or lacking experimental support. To demonstrate the applicability of the alchemical free energy method for protein–peptide systems in a typical real-world drug discovery project, we report an application of the thermodynamic integration (TI) method to the RBFE calculation of ghrelin receptor and its peptide agonists. Along with the calculation, the synthesis and in vitro EC50 activity of relamorelin and 17 new peptide derivatives were also reported. A cost-effective criterion to determine the data collection time was proposed for peptides in the TI simulation. The average of three TI repeats yielded a mean absolute error of 0.98 kcal/mol and Pearson’s correlation coefficient (R) of 0.77 against the experimental free energy derived from the in vitro EC50 activity, showing good repeatability of the proposed method and a slightly better agreement than the results obtained from the arbitrary time frames up to 20 ns. Although it is limited by having one target and a deduced binding pose, we hope that this study can add some insights into alchemical free energy calculation of protein–peptide systems, providing theoretical assistance to the development of peptide drugs.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
