Discovery of a novel BTK inhibitor S-016 and identification of a new strategy for the treatment of lymphomas including BTK inhibitor-resistant lymphomas.
Pei-Ran Song, Zhi-Peng Wan, Ge-Ge Huang, Zi-Lan Song, Tao Zhang, Lin-Jiang Tong, Yan Fang, Hao-Tian Tang, Yu Xue, Zheng-Sheng Zhan, Fang Feng, Yan Li, Wen-Hao Shi, Yu-Qing Huang, Yi Chen, Wen-Hu Duan, Jian Ding, Ao Zhang, Hua Xie
{"title":"Discovery of a novel BTK inhibitor S-016 and identification of a new strategy for the treatment of lymphomas including BTK inhibitor-resistant lymphomas.","authors":"Pei-Ran Song, Zhi-Peng Wan, Ge-Ge Huang, Zi-Lan Song, Tao Zhang, Lin-Jiang Tong, Yan Fang, Hao-Tian Tang, Yu Xue, Zheng-Sheng Zhan, Fang Feng, Yan Li, Wen-Hao Shi, Yu-Qing Huang, Yi Chen, Wen-Hu Duan, Jian Ding, Ao Zhang, Hua Xie","doi":"10.1038/s41401-024-01311-x","DOIUrl":null,"url":null,"abstract":"<p><p>Bruton's tyrosine kinase (BTK) has emerged as a therapeutic target for B-cell malignancies, which is substantiated by the efficacy of various irreversible or reversible BTK inhibitors. However, on-target BTK mutations facilitating evasion from BTK inhibition lead to resistance that limits the therapeutic efficacy of BTK inhibitors. In this study we employed structure-based drug design strategies based on established BTK inhibitors and yielded a series of BTK targeting compounds. Among them, compound S-016 bearing a unique tricyclic structure exhibited potent BTK kinase inhibitory activity with an IC<sub>50</sub> value of 0.5 nM, comparable to a commercially available BTK inhibitor ibrutinib (IC<sub>50</sub> = 0.4 nM). S-016, as a novel irreversible BTK inhibitor, displayed superior kinase selectivity compared to ibrutinib and significant therapeutic effects against B-cell lymphoma both in vitro and in vivo. Furthermore, we generated BTK inhibitor-resistant lymphoma cells harboring BTK C481F or A428D to explore strategies for overcoming resistance. Co-culture of these DLBCL cells with M0 macrophages led to the polarization of M0 macrophages toward the M2 phenotype, a process known to support tumor progression. Intriguingly, we demonstrated that SYHA1813, a compound targeting both VEGFR and CSF1R, effectively reshaped the tumor microenvironment (TME) and significantly overcame the acquired resistance to BTK inhibitors in both BTK-mutated and wild-type BTK DLBCL models by inhibiting angiogenesis and modulating macrophage polarization. Overall, this study not only promotes the development of new BTK inhibitors but also offers innovative treatment strategies for B-cell lymphomas, including those with BTK mutations.</p>","PeriodicalId":6942,"journal":{"name":"Acta Pharmacologica Sinica","volume":" ","pages":"2163-2173"},"PeriodicalIF":8.4000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11420226/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Pharmacologica Sinica","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41401-024-01311-x","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
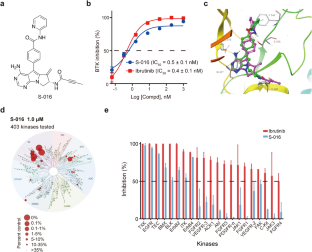
Bruton's tyrosine kinase (BTK) has emerged as a therapeutic target for B-cell malignancies, which is substantiated by the efficacy of various irreversible or reversible BTK inhibitors. However, on-target BTK mutations facilitating evasion from BTK inhibition lead to resistance that limits the therapeutic efficacy of BTK inhibitors. In this study we employed structure-based drug design strategies based on established BTK inhibitors and yielded a series of BTK targeting compounds. Among them, compound S-016 bearing a unique tricyclic structure exhibited potent BTK kinase inhibitory activity with an IC50 value of 0.5 nM, comparable to a commercially available BTK inhibitor ibrutinib (IC50 = 0.4 nM). S-016, as a novel irreversible BTK inhibitor, displayed superior kinase selectivity compared to ibrutinib and significant therapeutic effects against B-cell lymphoma both in vitro and in vivo. Furthermore, we generated BTK inhibitor-resistant lymphoma cells harboring BTK C481F or A428D to explore strategies for overcoming resistance. Co-culture of these DLBCL cells with M0 macrophages led to the polarization of M0 macrophages toward the M2 phenotype, a process known to support tumor progression. Intriguingly, we demonstrated that SYHA1813, a compound targeting both VEGFR and CSF1R, effectively reshaped the tumor microenvironment (TME) and significantly overcame the acquired resistance to BTK inhibitors in both BTK-mutated and wild-type BTK DLBCL models by inhibiting angiogenesis and modulating macrophage polarization. Overall, this study not only promotes the development of new BTK inhibitors but also offers innovative treatment strategies for B-cell lymphomas, including those with BTK mutations.
期刊介绍:
APS (Acta Pharmacologica Sinica) welcomes submissions from diverse areas of pharmacology and the life sciences. While we encourage contributions across a broad spectrum, topics of particular interest include, but are not limited to: anticancer pharmacology, cardiovascular and pulmonary pharmacology, clinical pharmacology, drug discovery, gastrointestinal and hepatic pharmacology, genitourinary, renal, and endocrine pharmacology, immunopharmacology and inflammation, molecular and cellular pharmacology, neuropharmacology, pharmaceutics, and pharmacokinetics. Join us in sharing your research and insights in pharmacology and the life sciences.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
