Nan Du, Xiaolei Wang, Zhaohui Wang, Hongwei Liu, Hui Liu, Hongfang Duan, Shaozhi Zhao, Santasree Banerjee, Xinwen Zhang
{"title":"Identification of a Novel Homozygous Mutation in <i>MTMR2</i> Gene Causes Very Rare Charcot-Marie-Tooth Disease Type 4B1.","authors":"Nan Du, Xiaolei Wang, Zhaohui Wang, Hongwei Liu, Hui Liu, Hongfang Duan, Shaozhi Zhao, Santasree Banerjee, Xinwen Zhang","doi":"10.2147/TACG.S448084","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Charcot-Marie-Tooth disease (CMT) is a heterogeneous group of disorders involving peripheral nervous system. Charcot-Marie-Tooth disease 4B1 (CMT4B1) is a rare subtype of CMT. CMT4B1 is an axonal demyelinating polyneuropathy with an autosomal recessive mode of inheritance. Patients with CMT4B1 usually manifested with dysfunction of the motor and sensory systems which leads to gradual and progressive muscular weakness and atrophy, starting from the peroneal muscles and finally affecting the distal muscles. Germline mutations in <i>MTMR2</i> gene causes CMT4B1.</p><p><strong>Material and methods: </strong>In this study, we investigated a 4-year-old Chinese boy with gradual and progressive weakness and atrophy of both proximal and distal muscles. The proband's parents did not show any abnormalities. Whole-exome sequencing and Sanger sequencing were performed.</p><p><strong>Results: </strong>Whole-exome sequencing identified a novel homozygous nonsense mutation (c.118A>T; p.Lys40*) in exon 2 of <i>MTMR2</i> gene in the proband. This novel mutation leads to the formation of a truncated MTMR2 protein of 39 amino acids instead of the wild- type MTMR2 protein of 643 amino acids. This mutation is predicted to cause the complete loss of the PH-GRAM domain, phosphatase domain, coiled-coil domain, and PDZ-binding motif of the MTMR2 protein. Sanger sequencing revealed that the proband's parents carried the mutation in a heterozygous state. This mutation was absent in 100 healthy control individuals.</p><p><strong>Conclusion: </strong>This study reports the first mutation in <i>MTMR2</i> associated with CMT4B1 in a Chinese population. Our study also showed the importance of whole-exome sequencing in identifying candidate genes and disease-causing variants in patients with CMT4B1.</p>","PeriodicalId":39131,"journal":{"name":"Application of Clinical Genetics","volume":"17 ","pages":"71-84"},"PeriodicalIF":2.6000,"publicationDate":"2024-05-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11149649/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S448084","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Charcot-Marie-Tooth disease (CMT) is a heterogeneous group of disorders involving peripheral nervous system. Charcot-Marie-Tooth disease 4B1 (CMT4B1) is a rare subtype of CMT. CMT4B1 is an axonal demyelinating polyneuropathy with an autosomal recessive mode of inheritance. Patients with CMT4B1 usually manifested with dysfunction of the motor and sensory systems which leads to gradual and progressive muscular weakness and atrophy, starting from the peroneal muscles and finally affecting the distal muscles. Germline mutations in MTMR2 gene causes CMT4B1.
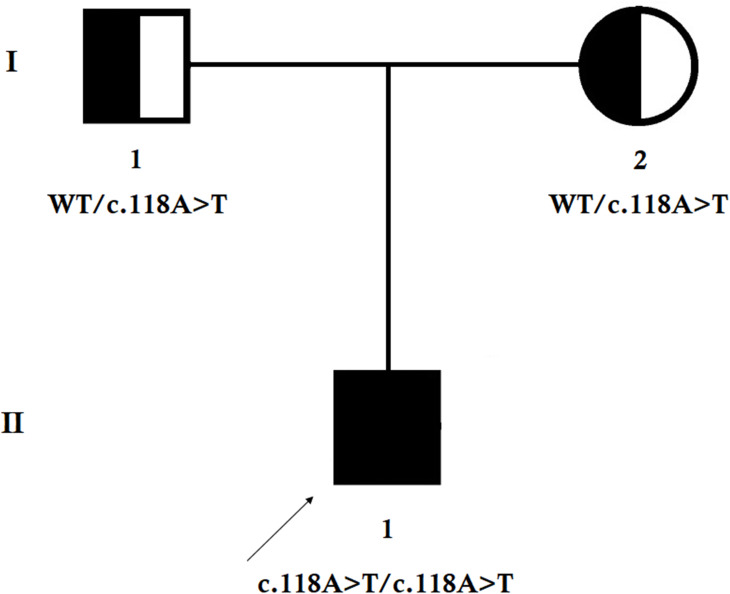
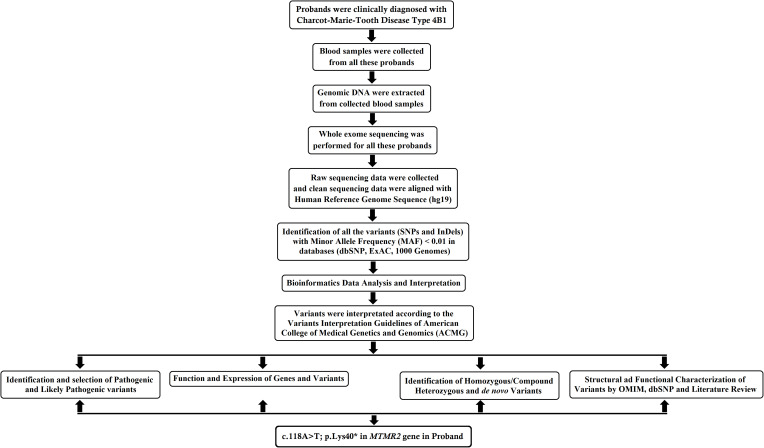
Material and methods: In this study, we investigated a 4-year-old Chinese boy with gradual and progressive weakness and atrophy of both proximal and distal muscles. The proband's parents did not show any abnormalities. Whole-exome sequencing and Sanger sequencing were performed.
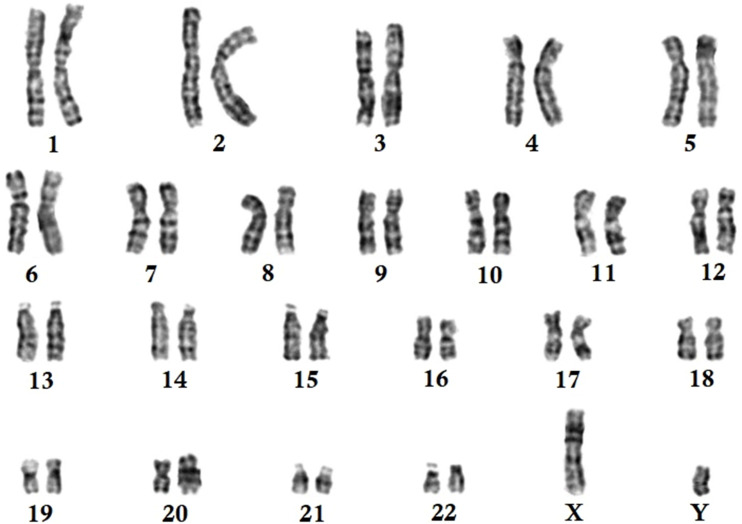
Results: Whole-exome sequencing identified a novel homozygous nonsense mutation (c.118A>T; p.Lys40*) in exon 2 of MTMR2 gene in the proband. This novel mutation leads to the formation of a truncated MTMR2 protein of 39 amino acids instead of the wild- type MTMR2 protein of 643 amino acids. This mutation is predicted to cause the complete loss of the PH-GRAM domain, phosphatase domain, coiled-coil domain, and PDZ-binding motif of the MTMR2 protein. Sanger sequencing revealed that the proband's parents carried the mutation in a heterozygous state. This mutation was absent in 100 healthy control individuals.
Conclusion: This study reports the first mutation in MTMR2 associated with CMT4B1 in a Chinese population. Our study also showed the importance of whole-exome sequencing in identifying candidate genes and disease-causing variants in patients with CMT4B1.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
