Brian L. Hie, Soochi Kim, Thomas A. Rando, Bryan Bryson, Bonnie Berger
{"title":"Scanorama: integrating large and diverse single-cell transcriptomic datasets","authors":"Brian L. Hie, Soochi Kim, Thomas A. Rando, Bryan Bryson, Bonnie Berger","doi":"10.1038/s41596-024-00991-3","DOIUrl":null,"url":null,"abstract":"Merging diverse single-cell RNA sequencing (scRNA-seq) data from numerous experiments, laboratories and technologies can uncover important biological insights. Nonetheless, integrating scRNA-seq data encounters special challenges when the datasets are composed of diverse cell type compositions. Scanorama offers a robust solution for improving the quality and interpretation of heterogeneous scRNA-seq data by effectively merging information from diverse sources. Scanorama is designed to address the technical variation introduced by differences in sample preparation, sequencing depth and experimental batches that can confound the analysis of multiple scRNA-seq datasets. Here we provide a detailed protocol for using Scanorama within a Scanpy-based single-cell analysis workflow coupled with Google Colaboratory, a cloud-based free Jupyter notebook environment service. The protocol involves Scanorama integration, a process that typically spans 0.5–3 h. Scanorama integration requires a basic understanding of cellular biology, transcriptomic technologies and bioinformatics. Our protocol and new Scanorama–Colaboratory resource should make scRNA-seq integration more widely accessible to researchers. Scanorama is an effective tool for combining multiple single-cell RNA sequencing datasets, addressing technical variation introduced by differences in sample preparation, sequencing depth and experimental batches that can confound the analysis of diverse datasets.","PeriodicalId":18901,"journal":{"name":"Nature Protocols","volume":"19 8","pages":"2283-2297"},"PeriodicalIF":16.0000,"publicationDate":"2024-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Protocols","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41596-024-00991-3","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
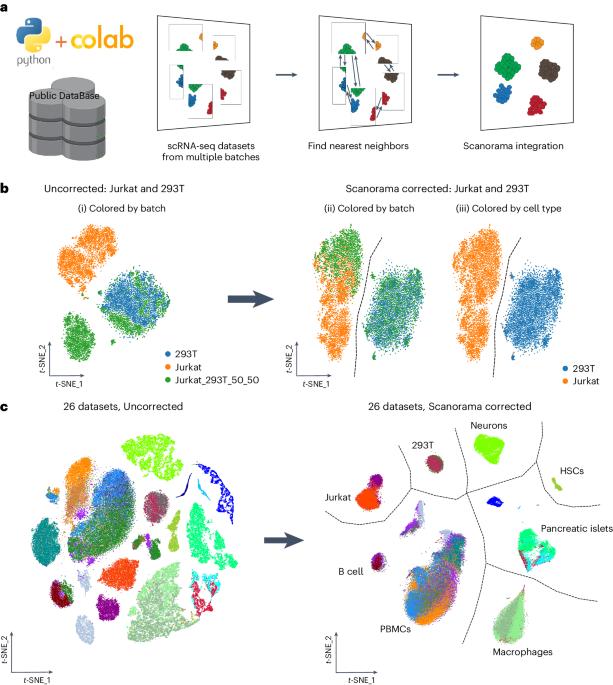
Merging diverse single-cell RNA sequencing (scRNA-seq) data from numerous experiments, laboratories and technologies can uncover important biological insights. Nonetheless, integrating scRNA-seq data encounters special challenges when the datasets are composed of diverse cell type compositions. Scanorama offers a robust solution for improving the quality and interpretation of heterogeneous scRNA-seq data by effectively merging information from diverse sources. Scanorama is designed to address the technical variation introduced by differences in sample preparation, sequencing depth and experimental batches that can confound the analysis of multiple scRNA-seq datasets. Here we provide a detailed protocol for using Scanorama within a Scanpy-based single-cell analysis workflow coupled with Google Colaboratory, a cloud-based free Jupyter notebook environment service. The protocol involves Scanorama integration, a process that typically spans 0.5–3 h. Scanorama integration requires a basic understanding of cellular biology, transcriptomic technologies and bioinformatics. Our protocol and new Scanorama–Colaboratory resource should make scRNA-seq integration more widely accessible to researchers. Scanorama is an effective tool for combining multiple single-cell RNA sequencing datasets, addressing technical variation introduced by differences in sample preparation, sequencing depth and experimental batches that can confound the analysis of diverse datasets.
期刊介绍:
Nature Protocols focuses on publishing protocols used to address significant biological and biomedical science research questions, including methods grounded in physics and chemistry with practical applications to biological problems. The journal caters to a primary audience of research scientists and, as such, exclusively publishes protocols with research applications. Protocols primarily aimed at influencing patient management and treatment decisions are not featured.
The specific techniques covered encompass a wide range, including but not limited to: Biochemistry, Cell biology, Cell culture, Chemical modification, Computational biology, Developmental biology, Epigenomics, Genetic analysis, Genetic modification, Genomics, Imaging, Immunology, Isolation, purification, and separation, Lipidomics, Metabolomics, Microbiology, Model organisms, Nanotechnology, Neuroscience, Nucleic-acid-based molecular biology, Pharmacology, Plant biology, Protein analysis, Proteomics, Spectroscopy, Structural biology, Synthetic chemistry, Tissue culture, Toxicology, and Virology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
