Siying Lin, Anthony G. Robson, Dorothy A. Thompson, Karolina M. Stepien, Robin Lachmann, Emma Footitt, Ola Czyz, Shwetha Chandrasekhar, Elena Schiff, Christos Iosifidis, Graeme C. Black, Michel Michaelides, Omar A. Mahroo, Gavin Arno, Andrew R. Webster
{"title":"Non-syndromic retinal dystrophy associated with biallelic variation of SUMF1 and reduced leukocyte sulfatase activity","authors":"Siying Lin, Anthony G. Robson, Dorothy A. Thompson, Karolina M. Stepien, Robin Lachmann, Emma Footitt, Ola Czyz, Shwetha Chandrasekhar, Elena Schiff, Christos Iosifidis, Graeme C. Black, Michel Michaelides, Omar A. Mahroo, Gavin Arno, Andrew R. Webster","doi":"10.1111/cge.14573","DOIUrl":null,"url":null,"abstract":"<p>Biallelic variants in <i>SUMF1</i> are associated with multiple sulfatase deficiency (MSD), a rare lysosomal storage disorder typically diagnosed in early infancy or childhood, marked by severe neurodegeneration and early mortality. We present clinical and molecular characterisation of three unrelated patients aged 13 to 58 years with milder clinical manifestations due to <i>SUMF1</i> disease variants, including two adult patients presenting with apparent non-syndromic retinal dystrophy. Whole genome sequencing identified biallelic <i>SUMF1</i> variants in all three patients; Patient 1 homozygous for a complex allele c.[290G>T;293T>A]; p.[(Gly97Val);(Val98Glu)], Patient 2 homozygous for c.866A>G; p.(Tyr289Cys), and Patient 3 compound heterozygous for c.726-1G>C and p.(Tyr289Cys). Electroretinography indicated a rod-cone dystrophy with additional possible inner retinal dysfunction in all three patients. Biochemical studies confirmed reduced, but not absent, sulfatase enzyme activity in the absence of extra-ocular disease (Patient 1) or only mild systemic disease (Patients 2, 3). These cases are suggestive that non-null <i>SUMF1</i> genotypes can cause an attenuated clinical phenotype, including retinal dystrophy without systemic complications, in adulthood.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 4","pages":"505-511"},"PeriodicalIF":2.3000,"publicationDate":"2024-06-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cge.14573","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14573","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
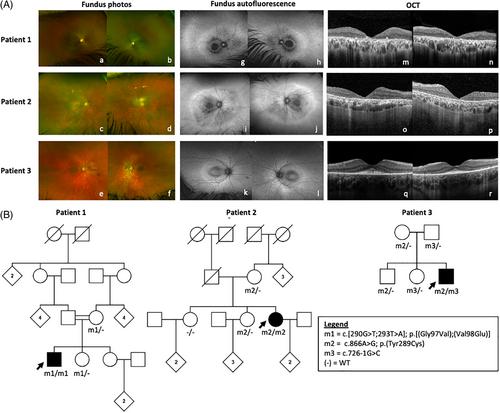
Biallelic variants in SUMF1 are associated with multiple sulfatase deficiency (MSD), a rare lysosomal storage disorder typically diagnosed in early infancy or childhood, marked by severe neurodegeneration and early mortality. We present clinical and molecular characterisation of three unrelated patients aged 13 to 58 years with milder clinical manifestations due to SUMF1 disease variants, including two adult patients presenting with apparent non-syndromic retinal dystrophy. Whole genome sequencing identified biallelic SUMF1 variants in all three patients; Patient 1 homozygous for a complex allele c.[290G>T;293T>A]; p.[(Gly97Val);(Val98Glu)], Patient 2 homozygous for c.866A>G; p.(Tyr289Cys), and Patient 3 compound heterozygous for c.726-1G>C and p.(Tyr289Cys). Electroretinography indicated a rod-cone dystrophy with additional possible inner retinal dysfunction in all three patients. Biochemical studies confirmed reduced, but not absent, sulfatase enzyme activity in the absence of extra-ocular disease (Patient 1) or only mild systemic disease (Patients 2, 3). These cases are suggestive that non-null SUMF1 genotypes can cause an attenuated clinical phenotype, including retinal dystrophy without systemic complications, in adulthood.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
