Rui Dong, Ruifeng Jin, Hongwei Zhang, Haiyan Zhang, Min Xue, Yue Li, Kaihui Zhang, Yuqiang Lv, Xiaoying Li, Yi Liu, Zhongtao Gai
{"title":"Genotypic and phenotypic characteristics of sodium channel—associated epilepsy in Chinese population","authors":"Rui Dong, Ruifeng Jin, Hongwei Zhang, Haiyan Zhang, Min Xue, Yue Li, Kaihui Zhang, Yuqiang Lv, Xiaoying Li, Yi Liu, Zhongtao Gai","doi":"10.1038/s10038-024-01257-2","DOIUrl":null,"url":null,"abstract":"Variants in voltage-gated sodium channel (VGSC) genes are implicated in seizures, epilepsy, and neurodevelopmental disorders, constituting a significant aspect of hereditary epilepsy in the Chinese population. Through retrospective analysis utilizing next-generation sequencing (NGS), we examined the genotypes and phenotypes of VGSC-related epilepsy cases from a cohort of 691 epilepsy subjects. Our findings revealed that 5.1% of subjects harbored VGSC variants, specifically 22 with SCN1A, 9 with SCN2A, 1 with SCN8A, and 3 with SCN1B variants; no SCN3A variants were detected. Among these, 14 variants were previously reported, while 21 were newly identified. SCN1A variant carriers predominantly presented with Dravet Syndrome (DS) and Genetic Epilepsy with Febrile Seizures Plus (GEFS + ), featuring a heightened sensitivity to fever-induced seizures. Statistically significant disparities emerged between the SCN1A-DS and SCN1A-GEFS+ groups concerning seizure onset and genetic diagnosis age, incidence of status epilepticus, mental retardation, anti-seizure medication (ASM) responsiveness, and familial history. Notably, subjects with SCN1A variants affecting the protein’s pore region experienced more frequent cluster seizures. All SCN2A variants were of de novo origin, and 88.9% of individuals with SCN2A variations exhibited cluster seizures. This research reveals a significant association between variations in VGSC-related genes and the clinical phenotype diversity of epilepsy subjects in China, emphasizing the pivotal role of NGS screening in establishing accurate disease diagnoses and guiding the selection of ASM.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 9","pages":"441-453"},"PeriodicalIF":2.5000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01257-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
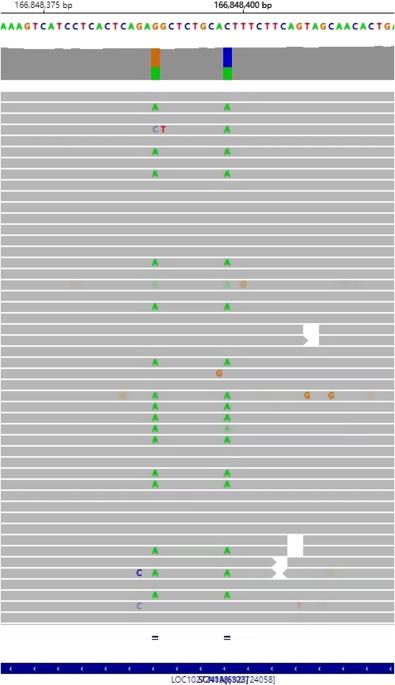
Variants in voltage-gated sodium channel (VGSC) genes are implicated in seizures, epilepsy, and neurodevelopmental disorders, constituting a significant aspect of hereditary epilepsy in the Chinese population. Through retrospective analysis utilizing next-generation sequencing (NGS), we examined the genotypes and phenotypes of VGSC-related epilepsy cases from a cohort of 691 epilepsy subjects. Our findings revealed that 5.1% of subjects harbored VGSC variants, specifically 22 with SCN1A, 9 with SCN2A, 1 with SCN8A, and 3 with SCN1B variants; no SCN3A variants were detected. Among these, 14 variants were previously reported, while 21 were newly identified. SCN1A variant carriers predominantly presented with Dravet Syndrome (DS) and Genetic Epilepsy with Febrile Seizures Plus (GEFS + ), featuring a heightened sensitivity to fever-induced seizures. Statistically significant disparities emerged between the SCN1A-DS and SCN1A-GEFS+ groups concerning seizure onset and genetic diagnosis age, incidence of status epilepticus, mental retardation, anti-seizure medication (ASM) responsiveness, and familial history. Notably, subjects with SCN1A variants affecting the protein’s pore region experienced more frequent cluster seizures. All SCN2A variants were of de novo origin, and 88.9% of individuals with SCN2A variations exhibited cluster seizures. This research reveals a significant association between variations in VGSC-related genes and the clinical phenotype diversity of epilepsy subjects in China, emphasizing the pivotal role of NGS screening in establishing accurate disease diagnoses and guiding the selection of ASM.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
