Aditya Kulkarni, Michael Bortz, Karl-Heinz Küfer, Maximilian Kohns* and Hans Hasse,
{"title":"Hierarchical Multicriteria Optimization of Molecular Models of Water","authors":"Aditya Kulkarni, Michael Bortz, Karl-Heinz Küfer, Maximilian Kohns* and Hans Hasse, ","doi":"10.1021/acs.jcim.4c00404","DOIUrl":null,"url":null,"abstract":"<p >Many widely used molecular models of water are built from a single Lennard-Jones site on which three point charges are positioned, one negative and two positive ones. Models from that class, denoted LJ3PC here, are computationally efficient, but it is well known that they cannot represent all relevant properties of water simultaneously with good accuracy. Despite the importance of the LJ3PC water model class, its inherent limitations in simultaneously describing different properties of water have never been studied systematically. This task can only be solved by multicriteria optimization (MCO). However, due to its computational cost, applying MCO to molecular models is a formidable task. We have recently introduced the reduced units method (RUM) to cope with this problem. In the present work, we apply the RUM in a hierarchical scheme to optimize LJ3PC water models taking into account five objectives: the representation of vapor pressure, saturated liquid density, self-diffusion coefficient, shear viscosity, and relative permittivity. Of the six parameters of the LJ3PC models, five were varied; only the H–O–H bond angle, which is usually chosen based on physical arguments, was kept constant. Our hierarchical RUM-based approach yields a Pareto set that contains attractive new water models. Furthermore, the results give an idea of what can be achieved by molecular modeling of water with models from the LJ3PC class.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"5077–5089"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00404","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
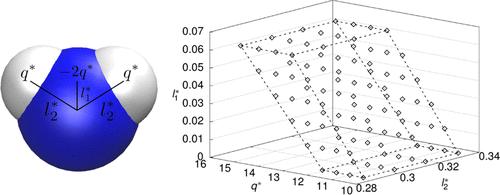
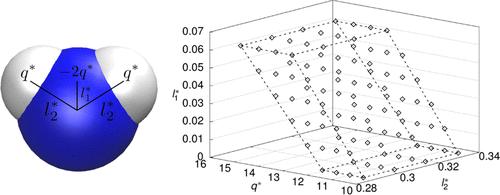
Many widely used molecular models of water are built from a single Lennard-Jones site on which three point charges are positioned, one negative and two positive ones. Models from that class, denoted LJ3PC here, are computationally efficient, but it is well known that they cannot represent all relevant properties of water simultaneously with good accuracy. Despite the importance of the LJ3PC water model class, its inherent limitations in simultaneously describing different properties of water have never been studied systematically. This task can only be solved by multicriteria optimization (MCO). However, due to its computational cost, applying MCO to molecular models is a formidable task. We have recently introduced the reduced units method (RUM) to cope with this problem. In the present work, we apply the RUM in a hierarchical scheme to optimize LJ3PC water models taking into account five objectives: the representation of vapor pressure, saturated liquid density, self-diffusion coefficient, shear viscosity, and relative permittivity. Of the six parameters of the LJ3PC models, five were varied; only the H–O–H bond angle, which is usually chosen based on physical arguments, was kept constant. Our hierarchical RUM-based approach yields a Pareto set that contains attractive new water models. Furthermore, the results give an idea of what can be achieved by molecular modeling of water with models from the LJ3PC class.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
