{"title":"Organic Synthesis Away from Equilibrium: Contrathermodynamic Transformations Enabled by Excited-State Electron Transfer","authors":"Angela Lin, Sumin Lee and Robert R. Knowles*, ","doi":"10.1021/acs.accounts.4c00227","DOIUrl":null,"url":null,"abstract":"<p >Chemists have long been inspired by biological photosynthesis, wherein a series of excited-state electron transfer (ET) events facilitate the conversion of low energy starting materials such as H<sub>2</sub>O and CO<sub>2</sub> into higher energy products in the form of carbohydrates and O<sub>2</sub>. While this model for utilizing light-driven charge transfer to drive catalytic reactions thermodynamically “uphill” has been extensively adapted for small molecule activation, molecular machines, photoswitches, and solar fuel chemistry, its application in organic synthesis has been less systematically developed. However, the potential benefits of these approaches are significant, both in enabling transformations that cannot be readily achieved using conventional thermal chemistry and in accessing distinct selectivity regimes that are uniquely enabled by excited-state mechanisms. In this Account, we present work from our group that highlights the ability of visible light photoredox catalysis to drive useful organic transformations away from their equilibrium positions, addressing a number of long-standing synthetic challenges.</p><p >We first discuss how excited-state ET enabled the first general methods for the catalytic anti-Markovnikov hydroamination of unactivated alkenes with alkyl amines. In these reactions, an excited-state iridium(III) photocatalyst reversibly oxidizes secondary amine substrates to their corresponding aminium radical cations (ARCs). These electrophilic <i>N</i>-centered radicals can then react with olefins to furnish valuable tertiary amine products with complete anti-Markovnikov regioselectivity. Notably, some of these products are less thermodynamically stable than their corresponding amine and alkene starting materials. We next present a strategy for light-driven C–C bond cleavage within various aliphatic alcohols mediated by homolytic activation of alcohol O–H bonds by excited-state proton-coupled electron transfer (PCET). The resulting alkoxy radical intermediates then undergo C–C β-scission to ultimately provide isomeric linear carbonyl products that are often higher in energy than their cyclic alcohol precursors. Applications of this chemistry for the light-driven depolymerization of lignin biomass, commercial phenoxy resin, hydroxylated polyolefin derivatives, and thermoset polymers are presented as well. We then describe a method for the contrathermodynamic positional isomerization of highly substituted olefins by means of cooperative photoredox and chromium(II) catalysis. In this work, generation of an allylchromium(III) species that can undergo highly regioselective <i>in situ</i> protodemetalation enables access to a less substituted and thermodynamically less stable positional isomer. Product selectivity in this reaction is determined by the large differential in oxidation potentials between differently substituted olefin isomers. Lastly, we discuss a light-driven deracemization reaction developed in collaboration with the Miller group, wherein a racemic urea substrate undergoes spontaneous optical enrichment upon visible light irradiation in the presence of an iridium(III) chromophore, a chiral Brønsted base, and a chiral peptide thiol. Excellent levels of enantioselectivity are achieved via sequential and synergistic proton transfer (PT) and H atom transfer (HAT) steps. Taken together, these examples highlight the ability of excited-state ET events to enable access to nonequilibrium product distributions across a wide range of catalytic, redox-neutral transformations in which photons are the only stoichiometric reagents.</p>","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":null,"pages":null},"PeriodicalIF":16.4000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.accounts.4c00227","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
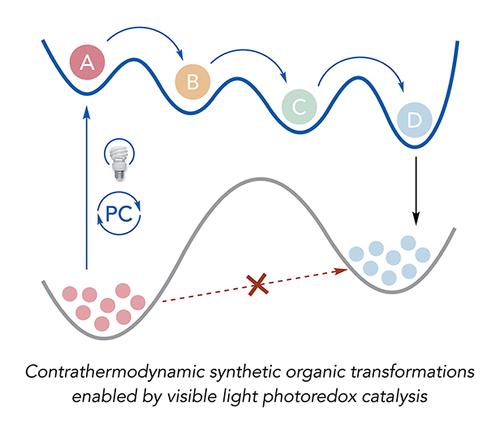
Chemists have long been inspired by biological photosynthesis, wherein a series of excited-state electron transfer (ET) events facilitate the conversion of low energy starting materials such as H2O and CO2 into higher energy products in the form of carbohydrates and O2. While this model for utilizing light-driven charge transfer to drive catalytic reactions thermodynamically “uphill” has been extensively adapted for small molecule activation, molecular machines, photoswitches, and solar fuel chemistry, its application in organic synthesis has been less systematically developed. However, the potential benefits of these approaches are significant, both in enabling transformations that cannot be readily achieved using conventional thermal chemistry and in accessing distinct selectivity regimes that are uniquely enabled by excited-state mechanisms. In this Account, we present work from our group that highlights the ability of visible light photoredox catalysis to drive useful organic transformations away from their equilibrium positions, addressing a number of long-standing synthetic challenges.
We first discuss how excited-state ET enabled the first general methods for the catalytic anti-Markovnikov hydroamination of unactivated alkenes with alkyl amines. In these reactions, an excited-state iridium(III) photocatalyst reversibly oxidizes secondary amine substrates to their corresponding aminium radical cations (ARCs). These electrophilic N-centered radicals can then react with olefins to furnish valuable tertiary amine products with complete anti-Markovnikov regioselectivity. Notably, some of these products are less thermodynamically stable than their corresponding amine and alkene starting materials. We next present a strategy for light-driven C–C bond cleavage within various aliphatic alcohols mediated by homolytic activation of alcohol O–H bonds by excited-state proton-coupled electron transfer (PCET). The resulting alkoxy radical intermediates then undergo C–C β-scission to ultimately provide isomeric linear carbonyl products that are often higher in energy than their cyclic alcohol precursors. Applications of this chemistry for the light-driven depolymerization of lignin biomass, commercial phenoxy resin, hydroxylated polyolefin derivatives, and thermoset polymers are presented as well. We then describe a method for the contrathermodynamic positional isomerization of highly substituted olefins by means of cooperative photoredox and chromium(II) catalysis. In this work, generation of an allylchromium(III) species that can undergo highly regioselective in situ protodemetalation enables access to a less substituted and thermodynamically less stable positional isomer. Product selectivity in this reaction is determined by the large differential in oxidation potentials between differently substituted olefin isomers. Lastly, we discuss a light-driven deracemization reaction developed in collaboration with the Miller group, wherein a racemic urea substrate undergoes spontaneous optical enrichment upon visible light irradiation in the presence of an iridium(III) chromophore, a chiral Brønsted base, and a chiral peptide thiol. Excellent levels of enantioselectivity are achieved via sequential and synergistic proton transfer (PT) and H atom transfer (HAT) steps. Taken together, these examples highlight the ability of excited-state ET events to enable access to nonequilibrium product distributions across a wide range of catalytic, redox-neutral transformations in which photons are the only stoichiometric reagents.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
