Yaqi Gao , Kyoungmin Kim , Heidi Vitrac , Rebecca L. Salazar , Benjamin D. Gould , Daniel Soedkamp , Weston Spivia , Koen Raedschelders , An Q. Dinh , Anna G. Guzman , Lin Tan , Stavros Azinas , David J.R. Taylor , Walter Schiffer , Daniel McNavish , Helen B. Burks , Roberta A. Gottlieb , Philip L. Lorenzi , Blake M. Hanson , Jennifer E. Van Eyk , Anja Karlstaedt
{"title":"Autophagic signaling promotes systems-wide remodeling in skeletal muscle upon oncometabolic stress by D2-HG","authors":"Yaqi Gao , Kyoungmin Kim , Heidi Vitrac , Rebecca L. Salazar , Benjamin D. Gould , Daniel Soedkamp , Weston Spivia , Koen Raedschelders , An Q. Dinh , Anna G. Guzman , Lin Tan , Stavros Azinas , David J.R. Taylor , Walter Schiffer , Daniel McNavish , Helen B. Burks , Roberta A. Gottlieb , Philip L. Lorenzi , Blake M. Hanson , Jennifer E. Van Eyk , Anja Karlstaedt","doi":"10.1016/j.molmet.2024.101969","DOIUrl":null,"url":null,"abstract":"<div><h3>Objectives</h3><p>Cachexia is a metabolic disorder and comorbidity with cancer and heart failure. The syndrome impacts more than thirty million people worldwide, accounting for 20% of all cancer deaths. In acute myeloid leukemia, somatic mutations of the metabolic enzyme isocitrate dehydrogenase 1 and 2 cause the production of the oncometabolite D2-hydroxyglutarate (D2-HG). Increased production of D2-HG is associated with heart and skeletal muscle atrophy, but the mechanistic links between metabolic and proteomic remodeling remain poorly understood. Therefore, we assessed how oncometabolic stress by D2-HG activates autophagy and drives skeletal muscle loss.</p></div><div><h3>Methods</h3><p>We quantified genomic, metabolomic, and proteomic changes in cultured skeletal muscle cells and mouse models of IDH-mutant leukemia using RNA sequencing, mass spectrometry, and computational modeling.</p></div><div><h3>Results</h3><p>D2-HG impairs NADH redox homeostasis in myotubes. Increased NAD+ levels drive activation of nuclear deacetylase Sirt1, which causes deacetylation and activation of LC3, a key regulator of autophagy. Using LC3 mutants, we confirm that deacetylation of LC3 by Sirt1 shifts its distribution from the nucleus into the cytosol, where it can undergo lipidation at pre-autophagic membranes. Sirt1 silencing or p300 overexpression attenuated autophagy activation in myotubes. <em>In vivo</em>, we identified increased muscle atrophy and reduced grip strength in response to D2-HG in male vs. female mice. In male mice, glycolytic intermediates accumulated, and protein expression of oxidative phosphorylation machinery was reduced. In contrast, female animals upregulated the same proteins, attenuating the phenotype <em>in vivo</em>. Network modeling and machine learning algorithms allowed us to identify candidate proteins essential for regulating oncometabolic adaptation in mouse skeletal muscle.</p></div><div><h3>Conclusions</h3><p>Our multi-omics approach exposes new metabolic vulnerabilities in response to D2-HG in skeletal muscle and provides a conceptual framework for identifying therapeutic targets in cachexia.</p></div>","PeriodicalId":18765,"journal":{"name":"Molecular Metabolism","volume":"86 ","pages":"Article 101969"},"PeriodicalIF":6.6000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2212877824001005/pdfft?md5=b088c88f4ae08c077b6c20892d34c2d8&pid=1-s2.0-S2212877824001005-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Metabolism","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2212877824001005","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives
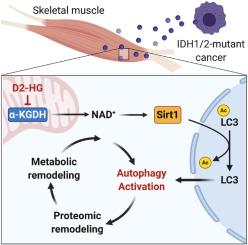
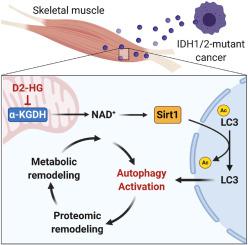
Cachexia is a metabolic disorder and comorbidity with cancer and heart failure. The syndrome impacts more than thirty million people worldwide, accounting for 20% of all cancer deaths. In acute myeloid leukemia, somatic mutations of the metabolic enzyme isocitrate dehydrogenase 1 and 2 cause the production of the oncometabolite D2-hydroxyglutarate (D2-HG). Increased production of D2-HG is associated with heart and skeletal muscle atrophy, but the mechanistic links between metabolic and proteomic remodeling remain poorly understood. Therefore, we assessed how oncometabolic stress by D2-HG activates autophagy and drives skeletal muscle loss.
Methods
We quantified genomic, metabolomic, and proteomic changes in cultured skeletal muscle cells and mouse models of IDH-mutant leukemia using RNA sequencing, mass spectrometry, and computational modeling.
Results
D2-HG impairs NADH redox homeostasis in myotubes. Increased NAD+ levels drive activation of nuclear deacetylase Sirt1, which causes deacetylation and activation of LC3, a key regulator of autophagy. Using LC3 mutants, we confirm that deacetylation of LC3 by Sirt1 shifts its distribution from the nucleus into the cytosol, where it can undergo lipidation at pre-autophagic membranes. Sirt1 silencing or p300 overexpression attenuated autophagy activation in myotubes. In vivo, we identified increased muscle atrophy and reduced grip strength in response to D2-HG in male vs. female mice. In male mice, glycolytic intermediates accumulated, and protein expression of oxidative phosphorylation machinery was reduced. In contrast, female animals upregulated the same proteins, attenuating the phenotype in vivo. Network modeling and machine learning algorithms allowed us to identify candidate proteins essential for regulating oncometabolic adaptation in mouse skeletal muscle.
Conclusions
Our multi-omics approach exposes new metabolic vulnerabilities in response to D2-HG in skeletal muscle and provides a conceptual framework for identifying therapeutic targets in cachexia.
期刊介绍:
Molecular Metabolism is a leading journal dedicated to sharing groundbreaking discoveries in the field of energy homeostasis and the underlying factors of metabolic disorders. These disorders include obesity, diabetes, cardiovascular disease, and cancer. Our journal focuses on publishing research driven by hypotheses and conducted to the highest standards, aiming to provide a mechanistic understanding of energy homeostasis-related behavior, physiology, and dysfunction.
We promote interdisciplinary science, covering a broad range of approaches from molecules to humans throughout the lifespan. Our goal is to contribute to transformative research in metabolism, which has the potential to revolutionize the field. By enabling progress in the prognosis, prevention, and ultimately the cure of metabolic disorders and their long-term complications, our journal seeks to better the future of health and well-being.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
