{"title":"The Conundrum of “Pair Sites” in Langmuir–Hinshelwood Reaction Kinetics in Heterogeneous Catalysis","authors":"Daniyal Kiani*, and , Israel E. Wachs*, ","doi":"10.1021/acscatal.4c02813","DOIUrl":null,"url":null,"abstract":"<p >Understanding reaction kinetics is crucial for designing and applying heterogeneous catalytic processes in chemical and energy conversion. Here, we revisit the Langmuir–Hinshelwood (L-H) kinetic model for bimolecular surface reactions, originally formulated for metal catalysts, assuming immobile adsorbates on neighboring pair sites, with the rate varying linearly with the density of surface sites (sites per unit area); <i>r</i> ∝ [*]<sub>o</sub><sup>1</sup>. Supported metal oxide catalysts, however, offer systematic control over [*]<sub>o</sub> through variation of the active two-dimensional metal oxide loading in the submonolayer region. Various reactions catalyzed by supported metal oxides are analyzed, such as supported VO<sub><i>x</i></sub> catalysts, including methanol oxidation, oxidative dehydrogenation of propane and ethane, SO<sub>2</sub> oxidation to SO<sub>3</sub>, propene oxidation to acrolein, <i>n</i>-butane oxidation to maleic anhydride, and selective catalytic reduction of nitric oxide with ammonia. The analysis reveals diverse dependencies of reaction rate on [*]<sub>o</sub> for these surface reactions, with <i>r</i> ∝ [*]<sub>o</sub><sup><i>n</i></sup>, where <i>n</i> equals 1 for reactions with a unimolecular rate-determining step and 2 for those with a bimolecular rate-limiting step or exchange of more than 2 electrons. We propose refraining from a priori assumptions about the nature and density of surface sites or adsorbate behavior, advocating instead for data-driven elucidation of kinetics based on the density of surface sites, adsorbate coverage, etc. Additionally, recent studies on catalytic surface mechanisms have shed light on nonadjacent catalytic sites catalyzing surface reactions in contrast to the traditional requirement of adjacent/pair sites. These findings underscore the need for a more nuanced approach in modeling heterogeneous catalysis, especially supported metal oxide catalysts, encouraging reliance on experimental data over idealized assumptions that are often difficult to justify.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":null,"pages":null},"PeriodicalIF":11.3000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acscatal.4c02813","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.4c02813","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
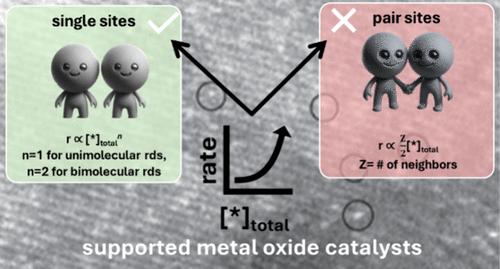
Understanding reaction kinetics is crucial for designing and applying heterogeneous catalytic processes in chemical and energy conversion. Here, we revisit the Langmuir–Hinshelwood (L-H) kinetic model for bimolecular surface reactions, originally formulated for metal catalysts, assuming immobile adsorbates on neighboring pair sites, with the rate varying linearly with the density of surface sites (sites per unit area); r ∝ [*]o1. Supported metal oxide catalysts, however, offer systematic control over [*]o through variation of the active two-dimensional metal oxide loading in the submonolayer region. Various reactions catalyzed by supported metal oxides are analyzed, such as supported VOx catalysts, including methanol oxidation, oxidative dehydrogenation of propane and ethane, SO2 oxidation to SO3, propene oxidation to acrolein, n-butane oxidation to maleic anhydride, and selective catalytic reduction of nitric oxide with ammonia. The analysis reveals diverse dependencies of reaction rate on [*]o for these surface reactions, with r ∝ [*]on, where n equals 1 for reactions with a unimolecular rate-determining step and 2 for those with a bimolecular rate-limiting step or exchange of more than 2 electrons. We propose refraining from a priori assumptions about the nature and density of surface sites or adsorbate behavior, advocating instead for data-driven elucidation of kinetics based on the density of surface sites, adsorbate coverage, etc. Additionally, recent studies on catalytic surface mechanisms have shed light on nonadjacent catalytic sites catalyzing surface reactions in contrast to the traditional requirement of adjacent/pair sites. These findings underscore the need for a more nuanced approach in modeling heterogeneous catalysis, especially supported metal oxide catalysts, encouraging reliance on experimental data over idealized assumptions that are often difficult to justify.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
