Hrishit Banerjee*, Clare P. Grey* and Andrew J. Morris*,
{"title":"Stability and Redox Mechanisms of Ni-Rich NMC Cathodes: Insights from First-Principles Many-Body Calculations","authors":"Hrishit Banerjee*, Clare P. Grey* and Andrew J. Morris*, ","doi":"10.1021/acs.chemmater.4c00928","DOIUrl":null,"url":null,"abstract":"<p >Ni-rich LiNi<sub><i>a</i></sub>Mn<sub><i>b</i></sub>Co<sub><i>c</i></sub>O<sub>2</sub> (NMC) cathodes undergo a series of degradation reactions, a prominent one being oxygen loss from the surface of the NMC particles; this process is more pronounced as Ni content is increased and at high voltages. Our first-principles study examines the redox behavior of transition metals (TMs) and O in Ni-rich NMC cathodes as a function of (de)lithiation. We use ab initio multiple scattering, density-functional theory (DFT)-based core-loss spectroscopy, and dynamical mean-field theory (DMFT) to give a many-body treatment of both dynamic and static correlations. Despite Ni, Mn, and Co K-edges calculated using ab initio multiple scattering based on Green’s functions showing an excellent match with experimentally obtained X-ray absorption near-edge spectra (XANES), we demonstrate that the ionic model of ascribing shifts in the XANES spectra to changes in metal oxidation states is inappropriate. We show that in these cases, which are characterized by strong covalency between the TM and oxygen, DMFT calculations based on Wannier projections are to date to the best of our knowledge the most accurate as well as computationally accessible method to calculate charges and hence assign oxidation states accurately. Due to the corresponding charge transfer from O p to Ni d, a ligand hole forms on O in Ni-rich regions. The individual Ni charge remains fairly constant throughout the charging/discharging process, particularly in Ni-rich environments in the material. In contrast, O has dual redox behavior, showing greater involvement in redox in Ni-rich regions while showing negligible redox involvement in Ni-poor regions. The Ni–O covalent system starts participating in redox around a state of delithiation of ∼17%, which represents, in our system, the beginning of the charge. Contrary to previous DFT calculations, we show that Co oxidation does not occur at the very end of charge but rather starts at an earlier state of delithiation of ∼67%. The dual behavior of O in terms of participation in the redox process helps explain the overall higher relative stability of lower Ni content NMCs compared to Ni-rich NMCs or LiNiO<sub>2</sub> in terms of O loss and evolution of singlet oxygen.</p>","PeriodicalId":33,"journal":{"name":"Chemistry of Materials","volume":"36 13","pages":"6575–6587"},"PeriodicalIF":7.0000,"publicationDate":"2024-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.chemmater.4c00928","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry of Materials","FirstCategoryId":"88","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.chemmater.4c00928","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
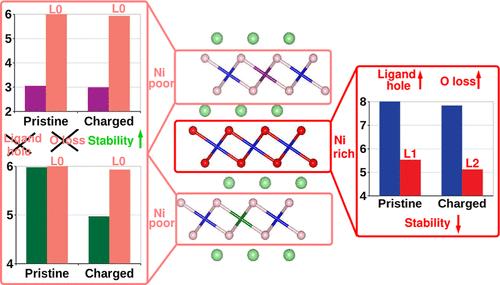
Ni-rich LiNiaMnbCocO2 (NMC) cathodes undergo a series of degradation reactions, a prominent one being oxygen loss from the surface of the NMC particles; this process is more pronounced as Ni content is increased and at high voltages. Our first-principles study examines the redox behavior of transition metals (TMs) and O in Ni-rich NMC cathodes as a function of (de)lithiation. We use ab initio multiple scattering, density-functional theory (DFT)-based core-loss spectroscopy, and dynamical mean-field theory (DMFT) to give a many-body treatment of both dynamic and static correlations. Despite Ni, Mn, and Co K-edges calculated using ab initio multiple scattering based on Green’s functions showing an excellent match with experimentally obtained X-ray absorption near-edge spectra (XANES), we demonstrate that the ionic model of ascribing shifts in the XANES spectra to changes in metal oxidation states is inappropriate. We show that in these cases, which are characterized by strong covalency between the TM and oxygen, DMFT calculations based on Wannier projections are to date to the best of our knowledge the most accurate as well as computationally accessible method to calculate charges and hence assign oxidation states accurately. Due to the corresponding charge transfer from O p to Ni d, a ligand hole forms on O in Ni-rich regions. The individual Ni charge remains fairly constant throughout the charging/discharging process, particularly in Ni-rich environments in the material. In contrast, O has dual redox behavior, showing greater involvement in redox in Ni-rich regions while showing negligible redox involvement in Ni-poor regions. The Ni–O covalent system starts participating in redox around a state of delithiation of ∼17%, which represents, in our system, the beginning of the charge. Contrary to previous DFT calculations, we show that Co oxidation does not occur at the very end of charge but rather starts at an earlier state of delithiation of ∼67%. The dual behavior of O in terms of participation in the redox process helps explain the overall higher relative stability of lower Ni content NMCs compared to Ni-rich NMCs or LiNiO2 in terms of O loss and evolution of singlet oxygen.
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
