{"title":"Childhood Cerebral Adrenoleukodystrophy: Case Report and Literature Review Advocating for Newborn Screening.","authors":"Hamrish Kumar Rajakumar, Varsha Coimbatore Sathyabal, Revathi Nachiappan, Sivakumar Krishnaswamy Vijayaramanujam","doi":"10.2147/DNND.S442985","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>X-linked adrenoleukodystrophy (ALD) is a rare genetic disorder caused by a pathogenic variant of the ABCD1 gene, leading to impaired peroxisomal function and the accumulation of very long-chain fatty acids (VLCFAs). ALD presents a wide range of neurological and adrenal symptoms, ranging from childhood cerebral adrenoleukodystrophy to adrenomyeloneuropathy and adrenal insufficiency. Newborn screening (NBS) for ALD is available in some regions but remains lacking in others, such as India.</p><p><strong>Case presentation: </strong>We present a case of a 10-year-old boy with ALD who presented with seizures, progressive weakness, visual impairment, and adrenal insufficiency. Despite symptomatic management and dietary adjustments, the disease progressed rapidly, leading to respiratory failure and eventual demise. The diagnosis was confirmed through molecular analysis and elevated VLCFA levels. Neuroimaging revealed characteristic white matter changes consistent with ALD.</p><p><strong>Conclusion: </strong>ALD is a devastating disease with no cure, emphasizing the importance of early detection through newborn screening and genetic testing. Management strategies include adrenal hormone therapy, gene therapy, and allogenic stem cell transplantation, as well as investigational treatments such as VLCFA normalization. Our case advocates the need for worldwide NBS and pediatric neurologic follow-up to enable early intervention and improve patient outcomes. Additionally, the association between ALD, recurrent febrile seizures, and unexplained developmental delay warrants further investigation to better understand disease progression and potential therapeutic targets.</p>","PeriodicalId":93972,"journal":{"name":"Degenerative neurological and neuromuscular disease","volume":"14 ","pages":"75-83"},"PeriodicalIF":3.2000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11192191/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Degenerative neurological and neuromuscular disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/DNND.S442985","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: X-linked adrenoleukodystrophy (ALD) is a rare genetic disorder caused by a pathogenic variant of the ABCD1 gene, leading to impaired peroxisomal function and the accumulation of very long-chain fatty acids (VLCFAs). ALD presents a wide range of neurological and adrenal symptoms, ranging from childhood cerebral adrenoleukodystrophy to adrenomyeloneuropathy and adrenal insufficiency. Newborn screening (NBS) for ALD is available in some regions but remains lacking in others, such as India.
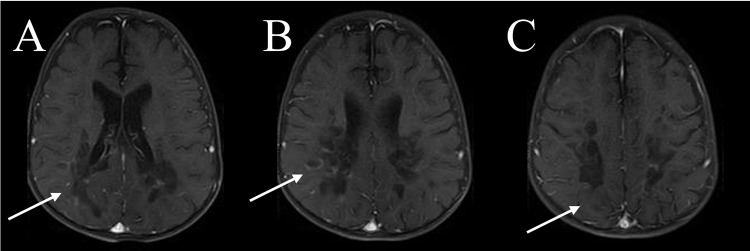
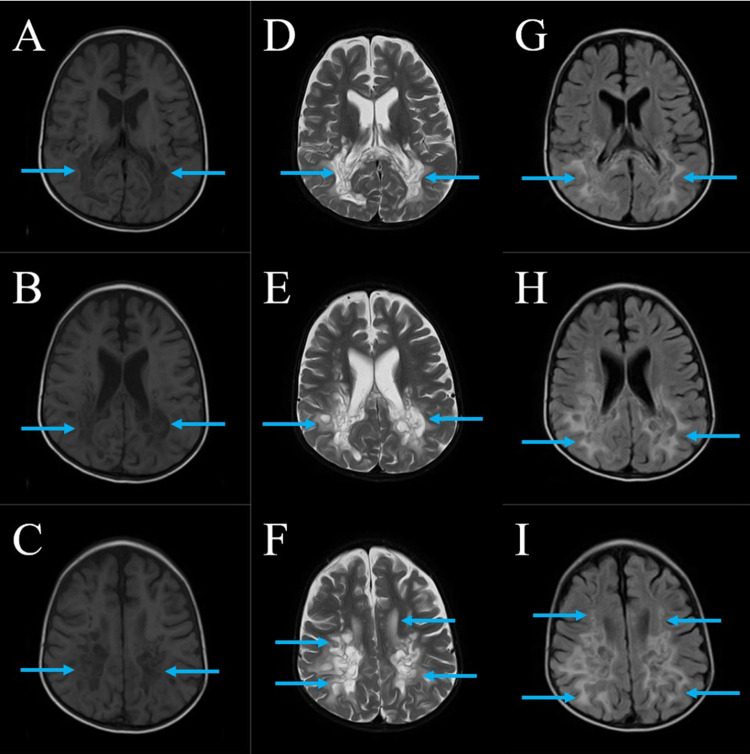
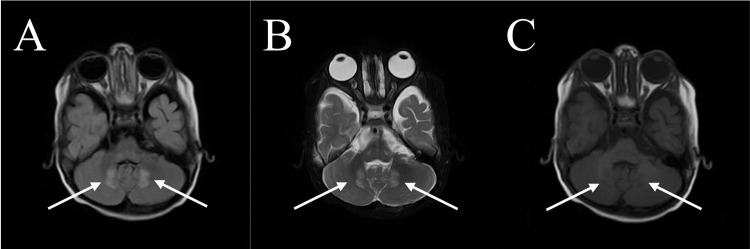
Case presentation: We present a case of a 10-year-old boy with ALD who presented with seizures, progressive weakness, visual impairment, and adrenal insufficiency. Despite symptomatic management and dietary adjustments, the disease progressed rapidly, leading to respiratory failure and eventual demise. The diagnosis was confirmed through molecular analysis and elevated VLCFA levels. Neuroimaging revealed characteristic white matter changes consistent with ALD.
Conclusion: ALD is a devastating disease with no cure, emphasizing the importance of early detection through newborn screening and genetic testing. Management strategies include adrenal hormone therapy, gene therapy, and allogenic stem cell transplantation, as well as investigational treatments such as VLCFA normalization. Our case advocates the need for worldwide NBS and pediatric neurologic follow-up to enable early intervention and improve patient outcomes. Additionally, the association between ALD, recurrent febrile seizures, and unexplained developmental delay warrants further investigation to better understand disease progression and potential therapeutic targets.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
