Kaname Kojima, Shu Tadaka, Yasunobu Okamura, Kengo Kinoshita
{"title":"Two-stage strategy using denoising autoencoders for robust reference-free genotype imputation with missing input genotypes","authors":"Kaname Kojima, Shu Tadaka, Yasunobu Okamura, Kengo Kinoshita","doi":"10.1038/s10038-024-01261-6","DOIUrl":null,"url":null,"abstract":"Widely used genotype imputation methods are based on the Li and Stephens model, which assumes that new haplotypes can be represented by modifying existing haplotypes in a reference panel through mutations and recombinations. These methods use genotypes from SNP arrays as inputs to estimate haplotypes that align with the input genotypes by analyzing recombination patterns within a reference panel, and then infer unobserved variants. While these methods require reference panels in an identifiable form, their public use is limited due to privacy and consent concerns. One strategy to overcome these limitations is to use de-identified haplotype information, such as summary statistics or model parameters. Advances in deep learning (DL) offer the potential to develop imputation methods that use haplotype information in a reference-free manner by handling it as model parameters, while maintaining comparable imputation accuracy to methods based on the Li and Stephens model. Here, we provide a brief introduction to DL-based reference-free genotype imputation methods, including RNN-IMP, developed by our research group. We then evaluate the performance of RNN-IMP against widely-used Li and Stephens model-based imputation methods in terms of accuracy (R2), using the 1000 Genomes Project Phase 3 dataset and corresponding simulated Omni2.5 SNP genotype data. Although RNN-IMP is sensitive to missing values in input genotypes, we propose a two-stage imputation strategy: missing genotypes are first imputed using denoising autoencoders; RNN-IMP then processes these imputed genotypes. This approach restores the imputation accuracy that is degraded by missing values, enhancing the practical use of RNN-IMP.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 10","pages":"511-518"},"PeriodicalIF":2.5000,"publicationDate":"2024-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01261-6.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01261-6","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
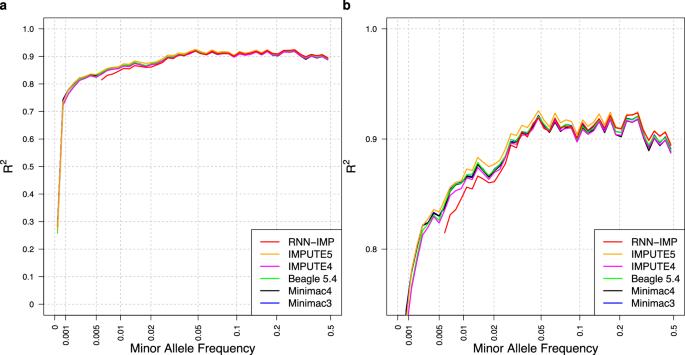
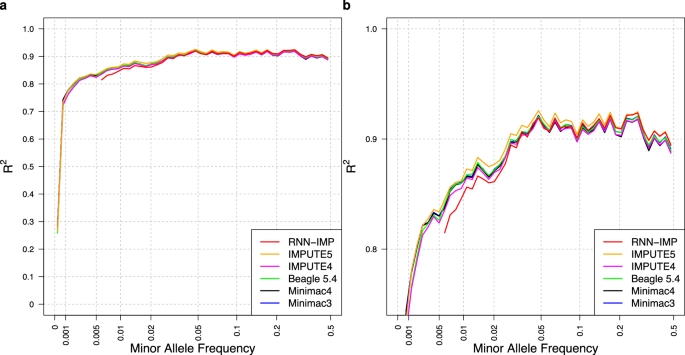
Widely used genotype imputation methods are based on the Li and Stephens model, which assumes that new haplotypes can be represented by modifying existing haplotypes in a reference panel through mutations and recombinations. These methods use genotypes from SNP arrays as inputs to estimate haplotypes that align with the input genotypes by analyzing recombination patterns within a reference panel, and then infer unobserved variants. While these methods require reference panels in an identifiable form, their public use is limited due to privacy and consent concerns. One strategy to overcome these limitations is to use de-identified haplotype information, such as summary statistics or model parameters. Advances in deep learning (DL) offer the potential to develop imputation methods that use haplotype information in a reference-free manner by handling it as model parameters, while maintaining comparable imputation accuracy to methods based on the Li and Stephens model. Here, we provide a brief introduction to DL-based reference-free genotype imputation methods, including RNN-IMP, developed by our research group. We then evaluate the performance of RNN-IMP against widely-used Li and Stephens model-based imputation methods in terms of accuracy (R2), using the 1000 Genomes Project Phase 3 dataset and corresponding simulated Omni2.5 SNP genotype data. Although RNN-IMP is sensitive to missing values in input genotypes, we propose a two-stage imputation strategy: missing genotypes are first imputed using denoising autoencoders; RNN-IMP then processes these imputed genotypes. This approach restores the imputation accuracy that is degraded by missing values, enhancing the practical use of RNN-IMP.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
