Josh Kemppainen*, Jacob R. Gissinger, S. Gowtham and Gregory M. Odegard,
{"title":"LUNAR: Automated Input Generation and Analysis for Reactive LAMMPS Simulations","authors":"Josh Kemppainen*, Jacob R. Gissinger, S. Gowtham and Gregory M. Odegard, ","doi":"10.1021/acs.jcim.4c00730","DOIUrl":null,"url":null,"abstract":"<p >Generating simulation-ready molecular models for the LAMMPS molecular dynamics (MD) simulation software package is a difficult task and impedes the more widespread and efficient use of MD in materials design and development. Fixed-bond force fields generally require manual assignment of atom types, bonded interactions, charges, and simulation domain sizes. A new LAMMPS pre- and postprocessing toolkit (LUNAR) is presented that efficiently builds molecular systems for LAMMPS. LUNAR automatically assigns atom types, generates bonded interactions, assigns charges, and provides initial configuration methods to generate large molecular systems. LUNAR can also incorporate chemical reactivity into simulations by facilitating the use of the REACTER protocol. Additionally, LUNAR provides postprocessing for free volume calculations, cure characterization calculations, and property predictions from LAMMPS thermodynamic outputs. LUNAR has been validated via building and simulation of pure epoxy and cyanate ester polymer systems with a comparison of the corresponding predicted structures and properties to benchmark values, including experimental results from the literature. LUNAR provides the tools for the computationally driven development of next-generation composite materials in the Integrated Computational Materials Engineering (ICME) and Materials Genome Initiative (MGI) frameworks. LUNAR is written in Python with the usage of NumPy and can be used via a graphical user interface, a command line interface, or an integrated design environment. LUNAR is freely available via GitHub.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"5108–5126"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00730","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00730","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
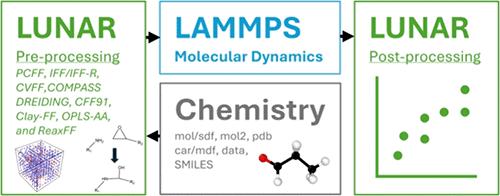
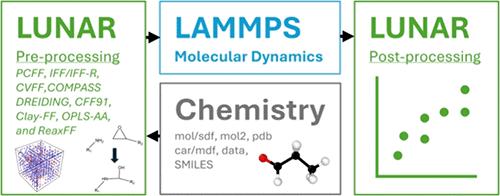
Generating simulation-ready molecular models for the LAMMPS molecular dynamics (MD) simulation software package is a difficult task and impedes the more widespread and efficient use of MD in materials design and development. Fixed-bond force fields generally require manual assignment of atom types, bonded interactions, charges, and simulation domain sizes. A new LAMMPS pre- and postprocessing toolkit (LUNAR) is presented that efficiently builds molecular systems for LAMMPS. LUNAR automatically assigns atom types, generates bonded interactions, assigns charges, and provides initial configuration methods to generate large molecular systems. LUNAR can also incorporate chemical reactivity into simulations by facilitating the use of the REACTER protocol. Additionally, LUNAR provides postprocessing for free volume calculations, cure characterization calculations, and property predictions from LAMMPS thermodynamic outputs. LUNAR has been validated via building and simulation of pure epoxy and cyanate ester polymer systems with a comparison of the corresponding predicted structures and properties to benchmark values, including experimental results from the literature. LUNAR provides the tools for the computationally driven development of next-generation composite materials in the Integrated Computational Materials Engineering (ICME) and Materials Genome Initiative (MGI) frameworks. LUNAR is written in Python with the usage of NumPy and can be used via a graphical user interface, a command line interface, or an integrated design environment. LUNAR is freely available via GitHub.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
