Pitcha Chompoopong MD, Michelle L. Mauermann MD, Hasan Siddiqi MD, Amanda Peltier MD
{"title":"Amyloid Neuropathy: From Pathophysiology to Treatment in Light-Chain Amyloidosis and Hereditary Transthyretin Amyloidosis","authors":"Pitcha Chompoopong MD, Michelle L. Mauermann MD, Hasan Siddiqi MD, Amanda Peltier MD","doi":"10.1002/ana.26965","DOIUrl":null,"url":null,"abstract":"<p>Amyloid neuropathy is caused by deposition of insoluble β-pleated amyloid sheets in the peripheral nervous system. It is most common in: (1) light-chain amyloidosis, a clonal non-proliferative plasma cell disorder in which fragments of immunoglobulin, light or heavy chain, deposit in tissues, and (2) hereditary transthyretin (ATTRv) amyloidosis, a disorder caused by autosomal dominant mutations in the <i>TTR</i> gene resulting in mutated protein that has a higher tendency to misfold. Amyloid fibrils deposit in the endoneurium of peripheral nerves, often extensive in the dorsal root ganglia and sympathetic ganglia, leading to atrophy of Schwann cells in proximity to amyloid fibrils and blood–nerve barrier disruption. Clinically, amyloid neuropathy is manifested as a length-dependent sensory predominant neuropathy associated with generalized autonomic failure. Small unmyelinated nerves are involved early and prominently in early-onset Val30Met ATTRv, whereas other ATTRv and light-chain amyloidosis often present with large- and small-fiber involvement. Nerve conduction studies, quantitative sudomotor axon testing, and intraepidermal nerve fiber density are useful tools to evaluate denervation. Amyloid deposition can be demonstrated by tissue biopsy of the affected organ or surrogate site, as well as bone-avid radiotracer cardiac imaging. Treatment of light-chain amyloidosis has been revolutionized by monoclonal antibodies and stem cell transplantation with improved 5-year survival up to 77%. Novel gene therapy and transthyretin stabilizers have revolutionized treatment of ATTRv, improving the course of neuropathy (less change in the modified Neuropathy Impairment Score + 7 from baseline) and quality of life. With great progress in amyloidosis therapies, early diagnosis and presymptomatic testing for ATTRv family members has become paramount. ANN NEUROL 2024;96:423–440</p>","PeriodicalId":127,"journal":{"name":"Annals of Neurology","volume":null,"pages":null},"PeriodicalIF":8.1000,"publicationDate":"2024-06-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ana.26965","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ana.26965","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
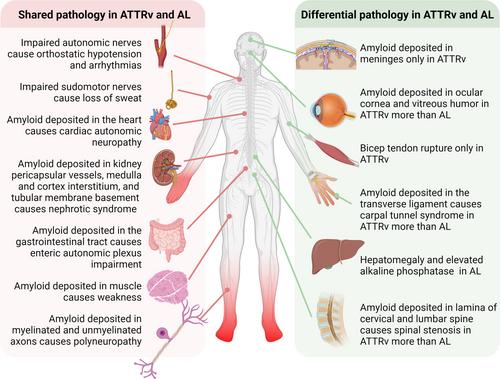
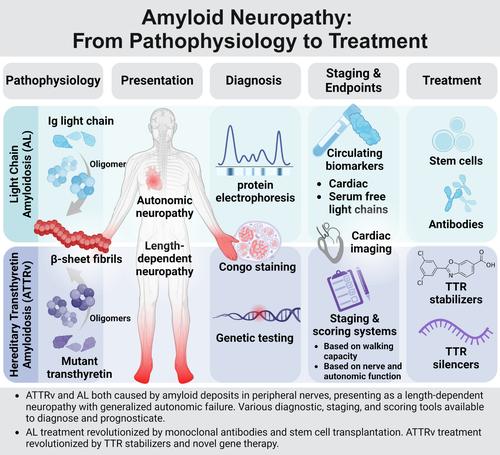
Amyloid neuropathy is caused by deposition of insoluble β-pleated amyloid sheets in the peripheral nervous system. It is most common in: (1) light-chain amyloidosis, a clonal non-proliferative plasma cell disorder in which fragments of immunoglobulin, light or heavy chain, deposit in tissues, and (2) hereditary transthyretin (ATTRv) amyloidosis, a disorder caused by autosomal dominant mutations in the TTR gene resulting in mutated protein that has a higher tendency to misfold. Amyloid fibrils deposit in the endoneurium of peripheral nerves, often extensive in the dorsal root ganglia and sympathetic ganglia, leading to atrophy of Schwann cells in proximity to amyloid fibrils and blood–nerve barrier disruption. Clinically, amyloid neuropathy is manifested as a length-dependent sensory predominant neuropathy associated with generalized autonomic failure. Small unmyelinated nerves are involved early and prominently in early-onset Val30Met ATTRv, whereas other ATTRv and light-chain amyloidosis often present with large- and small-fiber involvement. Nerve conduction studies, quantitative sudomotor axon testing, and intraepidermal nerve fiber density are useful tools to evaluate denervation. Amyloid deposition can be demonstrated by tissue biopsy of the affected organ or surrogate site, as well as bone-avid radiotracer cardiac imaging. Treatment of light-chain amyloidosis has been revolutionized by monoclonal antibodies and stem cell transplantation with improved 5-year survival up to 77%. Novel gene therapy and transthyretin stabilizers have revolutionized treatment of ATTRv, improving the course of neuropathy (less change in the modified Neuropathy Impairment Score + 7 from baseline) and quality of life. With great progress in amyloidosis therapies, early diagnosis and presymptomatic testing for ATTRv family members has become paramount. ANN NEUROL 2024;96:423–440
期刊介绍:
Annals of Neurology publishes original articles with potential for high impact in understanding the pathogenesis, clinical and laboratory features, diagnosis, treatment, outcomes and science underlying diseases of the human nervous system. Articles should ideally be of broad interest to the academic neurological community rather than solely to subspecialists in a particular field. Studies involving experimental model system, including those in cell and organ cultures and animals, of direct translational relevance to the understanding of neurological disease are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
