Magdalena W. Duszka, Michał F. Rode and Andrzej L. Sobolewski
{"title":"Computational design of boron-free triangular molecules with inverted singlet–triplet energy gap†","authors":"Magdalena W. Duszka, Michał F. Rode and Andrzej L. Sobolewski","doi":"10.1039/D4CP01658K","DOIUrl":null,"url":null,"abstract":"<p >A novel, computationally designed, class of triangular-shape organic molecules with an inverted singlet–triplet (IST) energy gap is investigated with <em>ab initio</em> electronic structure methods. The considered molecular systems are cyclic oligomers and their common feature is electronic conjugation along the molecular rim. Vertical excitation energies from the electronic ground state to the lowest singlet and triplet excited states were computed, as well as vertical emission energies from these states to the ground state. The results underscore the significance of optimizing excited-state geometries to accurately describe the optoelectronic properties of IST molecules, in particular with respect to their application in OLEDs.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 28","pages":" 19130-19137"},"PeriodicalIF":2.9000,"publicationDate":"2024-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01658k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
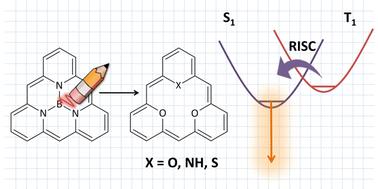
A novel, computationally designed, class of triangular-shape organic molecules with an inverted singlet–triplet (IST) energy gap is investigated with ab initio electronic structure methods. The considered molecular systems are cyclic oligomers and their common feature is electronic conjugation along the molecular rim. Vertical excitation energies from the electronic ground state to the lowest singlet and triplet excited states were computed, as well as vertical emission energies from these states to the ground state. The results underscore the significance of optimizing excited-state geometries to accurately describe the optoelectronic properties of IST molecules, in particular with respect to their application in OLEDs.
借助电子结构理论的 ab initio 方法,研究了一类通过计算设计的新型三角形有机分子,它们具有倒置的单电子-三电子(IST)能隙。所考虑的分子体系具有环状低聚物的形式,其共同特征是沿分子边缘局部的电子共轭。研究分析了所选分子从电子基态以及最低激发单重态和三重态的垂直转变能量。研究结果强调了在理论模型中优化激发态几何结构的重要性,以准确描述 IST 分子的光电特性,特别是与它们在有机发光二极管中的应用有关的特性。
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
