Gökhan Tahıl, Fabien Delorme, Daniel Le Berre, Éric Monflier, Adlane Sayede, Sébastien Tilloy
{"title":"Stereoisomers Are Not Machine Learning's Best Friends.","authors":"Gökhan Tahıl, Fabien Delorme, Daniel Le Berre, Éric Monflier, Adlane Sayede, Sébastien Tilloy","doi":"10.1021/acs.jcim.4c00318","DOIUrl":null,"url":null,"abstract":"<p><p>This study addresses the challenge of accurately identifying stereoisomers in cheminformatics, which originates from our objective to apply machine learning to predict the association constant between cyclodextrin and a guest. Identifying stereoisomers is indeed crucial for machine learning applications. Current tools offer various molecular descriptors, including their textual representation as Isomeric SMILES that can distinguish stereoisomers. However, such representation is text-based and does not have a fixed size, so a conversion is needed to make it usable to machine learning approaches. Word embedding techniques can be used to solve this problem. Mol2vec, a word embedding approach for molecules, offers such a conversion. Unfortunately, it cannot distinguish between stereoisomers due to its inability to capture the spatial configuration of molecular structures. This study proposes several approaches that use word embedding techniques to handle molecular discrimination using stereochemical information on molecules or considering Isomeric SMILES notation as a text in Natural Language Processing. Our aim is to generate a distinct vector for each unique molecule, correctly identifying stereoisomer information in cheminformatics. The proposed approaches are then compared to our original machine learning task: predicting the association constant between cyclodextrin and a guest molecule.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5451-5469"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00318","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
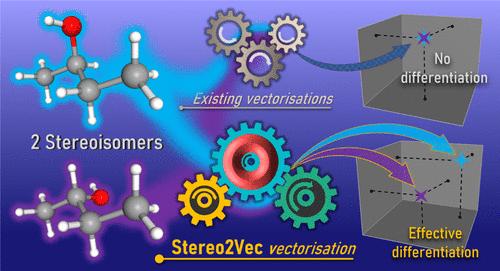
This study addresses the challenge of accurately identifying stereoisomers in cheminformatics, which originates from our objective to apply machine learning to predict the association constant between cyclodextrin and a guest. Identifying stereoisomers is indeed crucial for machine learning applications. Current tools offer various molecular descriptors, including their textual representation as Isomeric SMILES that can distinguish stereoisomers. However, such representation is text-based and does not have a fixed size, so a conversion is needed to make it usable to machine learning approaches. Word embedding techniques can be used to solve this problem. Mol2vec, a word embedding approach for molecules, offers such a conversion. Unfortunately, it cannot distinguish between stereoisomers due to its inability to capture the spatial configuration of molecular structures. This study proposes several approaches that use word embedding techniques to handle molecular discrimination using stereochemical information on molecules or considering Isomeric SMILES notation as a text in Natural Language Processing. Our aim is to generate a distinct vector for each unique molecule, correctly identifying stereoisomer information in cheminformatics. The proposed approaches are then compared to our original machine learning task: predicting the association constant between cyclodextrin and a guest molecule.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
