Jen-Tsan Chi, Chao Chieh Lin, Yi-Tzu Lin, Ssu-Yu Chen, Yasaman Setayeshpour, Yubin Chen, Denise Dunn, Erik Soderblom, Guo-Fang Zhang, Valeriy Filonenko, Suh Young Jeong, Scott Floyd, Susan Hayflick, Ivan Gout
{"title":"Coenzyme A protects against ferroptosis via CoAlation of thioredoxin reductase 2.","authors":"Jen-Tsan Chi, Chao Chieh Lin, Yi-Tzu Lin, Ssu-Yu Chen, Yasaman Setayeshpour, Yubin Chen, Denise Dunn, Erik Soderblom, Guo-Fang Zhang, Valeriy Filonenko, Suh Young Jeong, Scott Floyd, Susan Hayflick, Ivan Gout","doi":"10.21203/rs.3.rs-4522617/v1","DOIUrl":null,"url":null,"abstract":"<p><p>The Cystine-xCT transporter-Glutathione (GSH)-GPX4 axis is the canonical pathway to protect against ferroptosis. While not required for ferroptosis-inducing compounds (FINs) targeting GPX4, FINs targeting the xCT transporter require mitochondria and its lipid peroxidation to trigger ferroptosis. However, the mechanism underlying the difference between these FINs is still unknown. Given that cysteine is also required for coenzyme A (CoA) biosynthesis, here we show that CoA supplementation specifically prevents ferroptosis induced by xCT inhibitors but not GPX4 inhibitors. We find that, auranofin, a thioredoxin reductase inhibitor, abolishes the protective effect of CoA. We also find that CoA availability determines the enzymatic activity of thioredoxin reductase, but not thioredoxin. Importantly, the mitochondrial thioredoxin system, but not the cytosolic thioredoxin system, determines CoA-mediated ferroptosis inhibition. Our data show that the CoA regulates the <i>in vitro</i> enzymatic activity of mitochondrial thioredoxin reductase (TXNRD2) by covalently modifying the thiol group of cysteine (CoAlation) on Cys-483. Replacing Cys-483 with alanine on TXNRD2 abolishes its <i>in vitro</i> enzymatic activity and ability to protect cells from ferroptosis. Targeting xCT to limit cysteine import and, therefore, CoA biosynthesis reduced CoAlation on TXNRD2, an effect that was rescued by CoA supplementation. Furthermore, the fibroblasts from patients with disrupted CoA metabolism demonstrate increased mitochondrial lipid peroxidation. In organotypic brain slice cultures, inhibition of CoA biosynthesis leads to an oxidized thioredoxin system, mitochondrial lipid peroxidation, and loss in cell viability, which were all rescued by ferrostatin-1. These findings identify CoA-mediated post-translation modification to regulate the thioredoxin system as an alternative ferroptosis protection pathway with potential clinical relevance for patients with disrupted CoA metabolism.</p>","PeriodicalId":94282,"journal":{"name":"Research square","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11213209/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Research square","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.21203/rs.3.rs-4522617/v1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
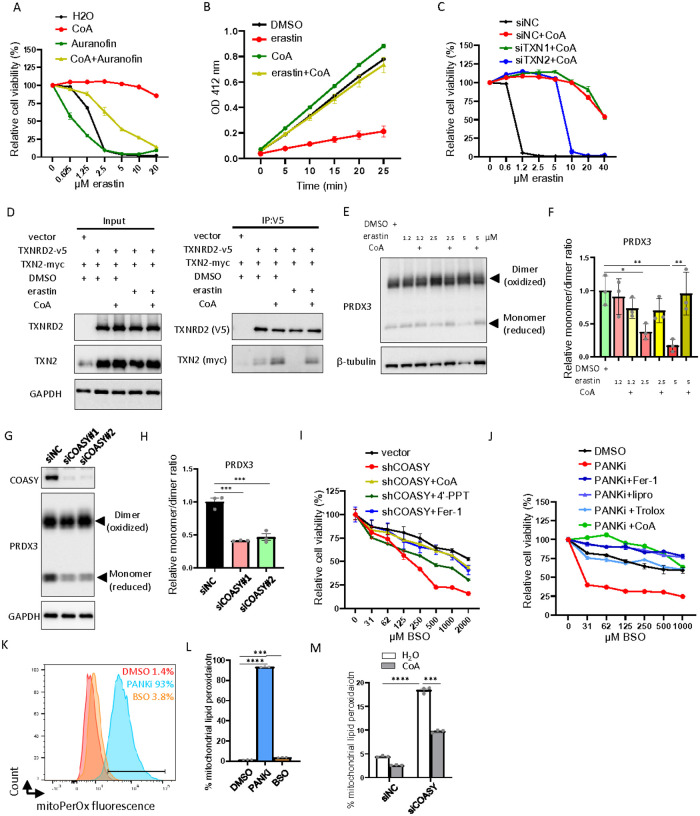
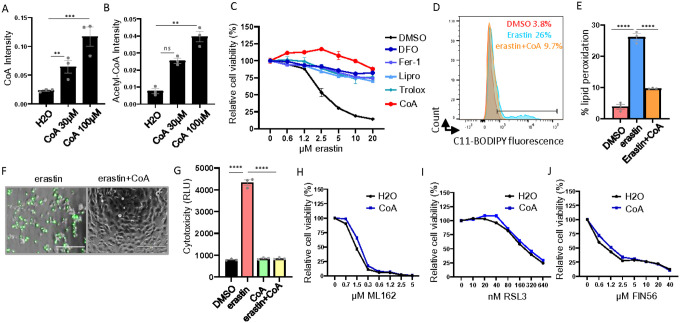
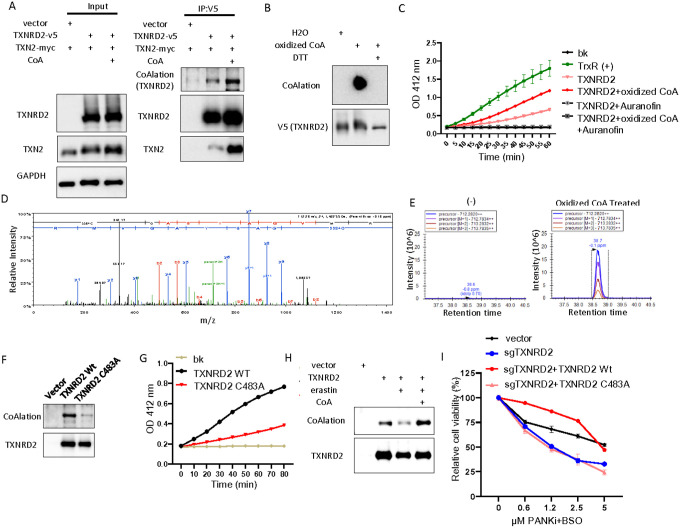
The Cystine-xCT transporter-Glutathione (GSH)-GPX4 axis is the canonical pathway to protect against ferroptosis. While not required for ferroptosis-inducing compounds (FINs) targeting GPX4, FINs targeting the xCT transporter require mitochondria and its lipid peroxidation to trigger ferroptosis. However, the mechanism underlying the difference between these FINs is still unknown. Given that cysteine is also required for coenzyme A (CoA) biosynthesis, here we show that CoA supplementation specifically prevents ferroptosis induced by xCT inhibitors but not GPX4 inhibitors. We find that, auranofin, a thioredoxin reductase inhibitor, abolishes the protective effect of CoA. We also find that CoA availability determines the enzymatic activity of thioredoxin reductase, but not thioredoxin. Importantly, the mitochondrial thioredoxin system, but not the cytosolic thioredoxin system, determines CoA-mediated ferroptosis inhibition. Our data show that the CoA regulates the in vitro enzymatic activity of mitochondrial thioredoxin reductase (TXNRD2) by covalently modifying the thiol group of cysteine (CoAlation) on Cys-483. Replacing Cys-483 with alanine on TXNRD2 abolishes its in vitro enzymatic activity and ability to protect cells from ferroptosis. Targeting xCT to limit cysteine import and, therefore, CoA biosynthesis reduced CoAlation on TXNRD2, an effect that was rescued by CoA supplementation. Furthermore, the fibroblasts from patients with disrupted CoA metabolism demonstrate increased mitochondrial lipid peroxidation. In organotypic brain slice cultures, inhibition of CoA biosynthesis leads to an oxidized thioredoxin system, mitochondrial lipid peroxidation, and loss in cell viability, which were all rescued by ferrostatin-1. These findings identify CoA-mediated post-translation modification to regulate the thioredoxin system as an alternative ferroptosis protection pathway with potential clinical relevance for patients with disrupted CoA metabolism.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
