Yu-Chi Kao, Yi-Ming Wang, Jyun-Yi Yeh, Shih-Cheng Li, Kevin C.-W. Wu, Li-Chiang Lin and Yi-Pei Li
{"title":"Tailoring parameters for QM/MM simulations: accurate modeling of adsorption and catalysis in zirconium-based metal–organic frameworks","authors":"Yu-Chi Kao, Yi-Ming Wang, Jyun-Yi Yeh, Shih-Cheng Li, Kevin C.-W. Wu, Li-Chiang Lin and Yi-Pei Li","doi":"10.1039/D4CP00681J","DOIUrl":null,"url":null,"abstract":"<p >Quantum mechanics/molecular mechanics (QM/MM) simulations offer an efficient way to model reactions occurring in complex environments. This study introduces a specialized set of charge and Lennard-Jones parameters tailored for electrostatically embedded QM/MM calculations, aiming to accurately model both adsorption processes and catalytic reactions in zirconium-based metal–organic frameworks (Zr-MOFs). To validate our approach, we compare adsorption energies derived from QM/MM simulations against experimental results and Monte Carlo simulation outcomes. The developed parameters showcase the ability of QM/MM simulations to represent long-range electrostatic and van der Waals interactions faithfully. This capability is evidenced by the prediction of adsorption energies with a low root mean square error of 1.1 kcal mol<small><sup>−1</sup></small> across a wide range of adsorbates. The practical applicability of our QM/MM model is further illustrated through the study of glucose isomerization and epimerization reactions catalyzed by two structurally distinct Zr-MOF catalysts, UiO-66 and MOF-808. Our QM/MM calculations closely align with experimental activation energies. Importantly, the parameter set introduced here is compatible with the widely used universal force field (UFF). Moreover, we thoroughly explore how the size of the cluster model and the choice of density functional theory (DFT) methodologies influence the simulation outcomes. This work provides an accurate and computationally efficient framework for modeling complex catalytic reactions within Zr-MOFs, contributing valuable insights into their mechanistic behaviors and facilitating further advancements in this dynamic area of research.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 30","pages":" 20388-20398"},"PeriodicalIF":2.9000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp00681j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
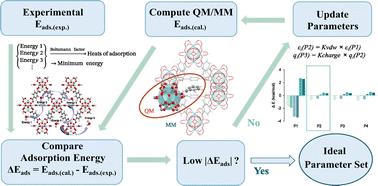
Quantum mechanics/molecular mechanics (QM/MM) simulations offer an efficient way to model reactions occurring in complex environments. This study introduces a specialized set of charge and Lennard-Jones parameters tailored for electrostatically embedded QM/MM calculations, aiming to accurately model both adsorption processes and catalytic reactions in zirconium-based metal–organic frameworks (Zr-MOFs). To validate our approach, we compare adsorption energies derived from QM/MM simulations against experimental results and Monte Carlo simulation outcomes. The developed parameters showcase the ability of QM/MM simulations to represent long-range electrostatic and van der Waals interactions faithfully. This capability is evidenced by the prediction of adsorption energies with a low root mean square error of 1.1 kcal mol−1 across a wide range of adsorbates. The practical applicability of our QM/MM model is further illustrated through the study of glucose isomerization and epimerization reactions catalyzed by two structurally distinct Zr-MOF catalysts, UiO-66 and MOF-808. Our QM/MM calculations closely align with experimental activation energies. Importantly, the parameter set introduced here is compatible with the widely used universal force field (UFF). Moreover, we thoroughly explore how the size of the cluster model and the choice of density functional theory (DFT) methodologies influence the simulation outcomes. This work provides an accurate and computationally efficient framework for modeling complex catalytic reactions within Zr-MOFs, contributing valuable insights into their mechanistic behaviors and facilitating further advancements in this dynamic area of research.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
