Paweł A. Wieczorkiewicz, Tadeusz M. Krygowski and Halina Szatylowicz
{"title":"Substituent effects and electron delocalization in five-membered N-heterocycles†","authors":"Paweł A. Wieczorkiewicz, Tadeusz M. Krygowski and Halina Szatylowicz","doi":"10.1039/D4CP01709A","DOIUrl":null,"url":null,"abstract":"<p >Five-membered N-heterocycles are principal constituents of many compounds of vital importance in various fields of chemistry, biochemistry or pharmaceutical chemistry. For this reason, unequivocal identification of structural factors determining electron donating/withdrawing properties of specific groups attached to the heterocyclic moiety becomes an utmost need together with elucidation of the substitution-induced changes in cyclic and noncyclic electron delocalization. Thus, quantum-chemical calculations were performed for pyrrole, imidazole, pyrazole, 1,2,3- and 1,2,4-triazole, and their C-substituted mono-derivatives (X = NO<small><sub>2</sub></small>, CN, Br, Cl, F, SH, OH, NH<small><sub>2</sub></small>). The obtained dataset contains information on substituent properties (cSAR – charge of the substituent active region method), delocalization (EDDB – electron density of delocalized bonds) and geometry. It follows that the positions of endocyclic N atoms relative to the substituent influence in the most profound manner its properties. N atoms in <em>ortho</em> positions significantly boost the electron-donation and weaken the electron-withdrawal by induction. Another factor is the resonance charge transfer from the substituents to N atoms, and then inductive interactions with further (non-<em>ortho</em>) N atoms. While substituent constants correctly describe the changes of their properties (including those attached to the heterocycles), a testimony to Hammett's genius, quantum chemical models must be used to quantify the exact properties. In most heterocycles, electron-donating substituents hinder the cyclic delocalization, except 4-pyrazole. The applied recent EDDB method allows to study this phenomenon in detail. It follows that changes in aromaticity originate from the π-electronic effects of substituents on the ring bonds, changing the localization and delocalization of particular bonds in a correlated manner.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 28","pages":" 19398-19410"},"PeriodicalIF":2.9000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp01709a?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01709a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
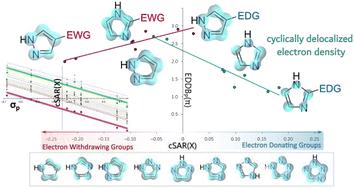
Five-membered N-heterocycles are principal constituents of many compounds of vital importance in various fields of chemistry, biochemistry or pharmaceutical chemistry. For this reason, unequivocal identification of structural factors determining electron donating/withdrawing properties of specific groups attached to the heterocyclic moiety becomes an utmost need together with elucidation of the substitution-induced changes in cyclic and noncyclic electron delocalization. Thus, quantum-chemical calculations were performed for pyrrole, imidazole, pyrazole, 1,2,3- and 1,2,4-triazole, and their C-substituted mono-derivatives (X = NO2, CN, Br, Cl, F, SH, OH, NH2). The obtained dataset contains information on substituent properties (cSAR – charge of the substituent active region method), delocalization (EDDB – electron density of delocalized bonds) and geometry. It follows that the positions of endocyclic N atoms relative to the substituent influence in the most profound manner its properties. N atoms in ortho positions significantly boost the electron-donation and weaken the electron-withdrawal by induction. Another factor is the resonance charge transfer from the substituents to N atoms, and then inductive interactions with further (non-ortho) N atoms. While substituent constants correctly describe the changes of their properties (including those attached to the heterocycles), a testimony to Hammett's genius, quantum chemical models must be used to quantify the exact properties. In most heterocycles, electron-donating substituents hinder the cyclic delocalization, except 4-pyrazole. The applied recent EDDB method allows to study this phenomenon in detail. It follows that changes in aromaticity originate from the π-electronic effects of substituents on the ring bonds, changing the localization and delocalization of particular bonds in a correlated manner.
五元 N-杂环是许多化合物的主要成分,在化学、生物化学或药物化学的各个领域都具有重要意义。因此,明确鉴定决定杂环分子上特定基团电子捐赠/抽取特性的结构因素,以及阐明取代引起的环状和非环状电子脱定位变化,已成为当务之急。因此,我们对吡咯、咪唑、吡唑、1,2,3- 和 1,2,4-三唑及其 C 取代单衍生物(X = NO2、CN、Br、Cl、F、SH、OH、NH2)进行了量子化学计算。所获得的数据集包含有关取代基性质(cSAR--取代基活性区电荷法)、脱局(EDB--脱局键电子密度)和几何形状的信息。由此可见,内环 N 原子相对于取代基的位置对其性质影响最大。位于正交位置的 N 原子通过诱导作用显著增强了电子捐赠作用,削弱了电子撤回作用。另一个因素是从取代基到 N 原子的共振电荷转移,然后与更多的(非正交)N 原子发生感应作用。虽然取代基常数能正确描述其性质(包括杂环上的取代基常数)的变化,证明了哈米特的天才,但量子化学模型必须用来量化确切的性质。在大多数杂环中,除 4-吡唑外,电子捐赠取代基都会阻碍环向脱钙化。最近应用的 EDDB 方法可以详细研究这一现象。由此可见,芳香性的变化源于取代基对环键的π电子效应,它以相关的方式改变了特定键的定位和脱定位。
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
