{"title":"Paramagnetic Properties of [An<sup>IV</sup>(NO<sub>3</sub>)<sub>6</sub>]<sup>2-</sup> Complexes (An = U, Np, Pu) Probed by NMR Spectroscopy and Quantum Chemical Calculations.","authors":"Matthieu Autillo, Marie-Claire Illy, Luca Briscese, Md Ashraful Islam, Hélène Bolvin, Claude Berthon","doi":"10.1021/acs.inorgchem.4c01694","DOIUrl":null,"url":null,"abstract":"<p><p>Actinide +IV complexes with six nitrates [An<sup>IV</sup>(NO<sub>3</sub>)<sub>6</sub>]<sup>2-</sup> (An = Th, U, Np, and Pu) have been studied by <sup>15</sup>N and <sup>17</sup>O NMR spectroscopy in solution and first-principles calculations. Magnetic susceptibilities were evaluated experimentally using the Evans method and are in good agreement with the ab initio values. The evolution in the series of the crystal field parameters deduced from ab initio calculations is discussed. The NMR paramagnetic shifts are analyzed based on ab initio calculations. Because the cubic symmetry of the complex quenches the dipolar contribution, they are only of Fermi contact origin. They are evaluated from first-principles based on a complete active space/density functional theory (DFT) strategy, in good accordance with the experimental one. The ligand hyperfine coupling constants are deduced from paramagnetic shifts and calculated using unrestricted DFT. The latter are decomposed in terms of the contribution of molecular orbitals. It highlights two pathways for the delocalization of the spin density from the metallic open-shell 5f orbitals to the NMR active nuclei, either through the valence 5f hybridized with 6d to the valence 2p molecular orbitals of the ligands, or by spin polarization of the metallic 6p orbitals which interact with the 2s-based molecular orbitals of the ligands.</p>","PeriodicalId":40,"journal":{"name":"Inorganic Chemistry","volume":null,"pages":null},"PeriodicalIF":4.3000,"publicationDate":"2024-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.inorgchem.4c01694","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
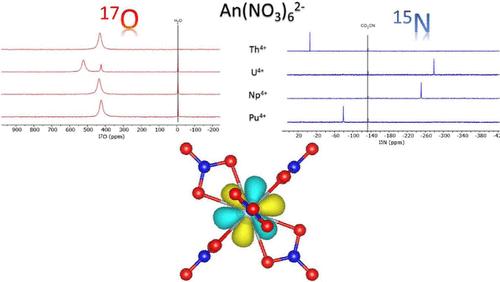
Abstract
Actinide +IV complexes with six nitrates [AnIV(NO3)6]2- (An = Th, U, Np, and Pu) have been studied by 15N and 17O NMR spectroscopy in solution and first-principles calculations. Magnetic susceptibilities were evaluated experimentally using the Evans method and are in good agreement with the ab initio values. The evolution in the series of the crystal field parameters deduced from ab initio calculations is discussed. The NMR paramagnetic shifts are analyzed based on ab initio calculations. Because the cubic symmetry of the complex quenches the dipolar contribution, they are only of Fermi contact origin. They are evaluated from first-principles based on a complete active space/density functional theory (DFT) strategy, in good accordance with the experimental one. The ligand hyperfine coupling constants are deduced from paramagnetic shifts and calculated using unrestricted DFT. The latter are decomposed in terms of the contribution of molecular orbitals. It highlights two pathways for the delocalization of the spin density from the metallic open-shell 5f orbitals to the NMR active nuclei, either through the valence 5f hybridized with 6d to the valence 2p molecular orbitals of the ligands, or by spin polarization of the metallic 6p orbitals which interact with the 2s-based molecular orbitals of the ligands.
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
