Benjamin Ries, Irfan Alibay, Nithishwer Mouroug Anand, Philip C Biggin, Aniket Magarkar
{"title":"Automated Absolute Binding Free Energy Calculation Workflow for Drug Discovery.","authors":"Benjamin Ries, Irfan Alibay, Nithishwer Mouroug Anand, Philip C Biggin, Aniket Magarkar","doi":"10.1021/acs.jcim.4c00343","DOIUrl":null,"url":null,"abstract":"<p><p>Absolute binding free energies play a crucial role in drug development, particularly as part of the lead discovery process. In recent work, we showed how <i>in silico</i> predictions directly could support drug development by ranking and recommending favorable ideas over unfavorable ones. Here, we demonstrate a Python workflow that enables the calculation of ABFEs with minimal manual input effort, such as the receptor PDB and ligand SDF files, and outputs a .tsv file containing the ranked ligands and their corresponding binding free energies. The implementation uses Snakemake to structure and control the execution of tasks, allowing for dynamic control of parameters and execution patterns. We provide an example of a benchmark system that demonstrates the effectiveness of the automated workflow.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5357-5364"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00343","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
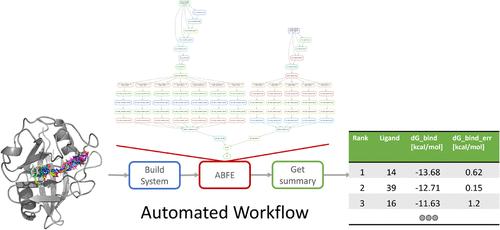
Absolute binding free energies play a crucial role in drug development, particularly as part of the lead discovery process. In recent work, we showed how in silico predictions directly could support drug development by ranking and recommending favorable ideas over unfavorable ones. Here, we demonstrate a Python workflow that enables the calculation of ABFEs with minimal manual input effort, such as the receptor PDB and ligand SDF files, and outputs a .tsv file containing the ranked ligands and their corresponding binding free energies. The implementation uses Snakemake to structure and control the execution of tasks, allowing for dynamic control of parameters and execution patterns. We provide an example of a benchmark system that demonstrates the effectiveness of the automated workflow.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
