{"title":"Scaffold-Hopped Compound Identification by Ligand-Based Approaches with a Prospective Affinity Test.","authors":"Itsuki Maeda, Shunsuke Tamura, Yoshihiro Ogura, Takayuki Serizawa, Takashi Shimada, Ryo Kunimoto, Tomoyuki Miyao","doi":"10.1021/acs.jcim.4c00342","DOIUrl":null,"url":null,"abstract":"<p><p>Scaffold-hopped (SH) compounds are bioactive compounds structurally different from known active compounds. Identifying SH compounds in the ligand-based approaches has been a central issue in medicinal chemistry, and various molecular representations of scaffold hopping have been proposed. However, appropriate representations for SH compound identification remain unclear. Herein, the ability of SH compound identification among several representations was fairly evaluated based on retrospective validation and prospective demonstration. In the retrospective validation, the combinations of two screening algorithms and four two- and three-dimensional molecular representations were compared using controlled data sets for the early identification of SH compounds. We found that the combination of the support vector machine and extended connectivity fingerprint with bond diameter 4 (SVM-ECFP4) and SVM and the rapid overlay of chemical structures (SVM-ROCS) showed a relatively high performance. The compounds that were highly ranked by SVM-ROCS did not share substructures with the active training compounds, while those ranked by SVM-ECFP4 were mostly recombinant. In the prospective demonstration, 93 SH compounds were prepared by screening the Namiki database using SVM-ROCS, targeting ABL1 inhibitors. The primary screening using surface plasmon resonance suggested five active compounds; however, in the competitive binding assays with adenosine triphosphate, no hits were found.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5557-5569"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267578/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00342","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
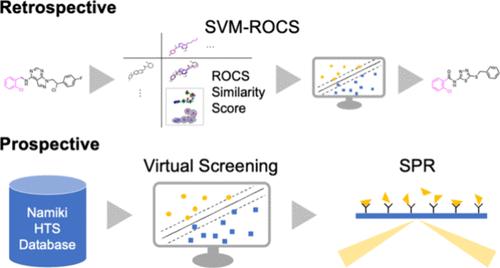
Scaffold-hopped (SH) compounds are bioactive compounds structurally different from known active compounds. Identifying SH compounds in the ligand-based approaches has been a central issue in medicinal chemistry, and various molecular representations of scaffold hopping have been proposed. However, appropriate representations for SH compound identification remain unclear. Herein, the ability of SH compound identification among several representations was fairly evaluated based on retrospective validation and prospective demonstration. In the retrospective validation, the combinations of two screening algorithms and four two- and three-dimensional molecular representations were compared using controlled data sets for the early identification of SH compounds. We found that the combination of the support vector machine and extended connectivity fingerprint with bond diameter 4 (SVM-ECFP4) and SVM and the rapid overlay of chemical structures (SVM-ROCS) showed a relatively high performance. The compounds that were highly ranked by SVM-ROCS did not share substructures with the active training compounds, while those ranked by SVM-ECFP4 were mostly recombinant. In the prospective demonstration, 93 SH compounds were prepared by screening the Namiki database using SVM-ROCS, targeting ABL1 inhibitors. The primary screening using surface plasmon resonance suggested five active compounds; however, in the competitive binding assays with adenosine triphosphate, no hits were found.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
