Michael Vacher, Rodrigo Canovas, Simon M Laws, James D Doecke
{"title":"A comprehensive multi-omics analysis reveals unique signatures to predict Alzheimer's disease.","authors":"Michael Vacher, Rodrigo Canovas, Simon M Laws, James D Doecke","doi":"10.3389/fbinf.2024.1390607","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Complex disorders, such as Alzheimer's disease (AD), result from the combined influence of multiple biological and environmental factors. The integration of high-throughput data from multiple omics platforms can provide system overviews, improving our understanding of complex biological processes underlying human disease. In this study, integrated data from four omics platforms were used to characterise biological signatures of AD.</p><p><strong>Method: </strong>The study cohort consists of 455 participants (Control:148, Cases:307) from the Religious Orders Study and Memory and Aging Project (ROSMAP). Genotype (SNP), methylation (CpG), RNA and proteomics data were collected, quality-controlled and pre-processed (SNP = 130; CpG = 83; RNA = 91; Proteomics = 119). Using a diagnosis of Mild Cognitive Impairment (MCI)/AD combined as the target phenotype, we first used Partial Least Squares Regression as an unsupervised classification framework to assess the prediction capabilities for each omics dataset individually. We then used a variation of the sparse generalized canonical correlation analysis (sGCCA) to assess predictions of the combined datasets and identify multi-omics signatures characterising each group of participants.</p><p><strong>Results: </strong>Analysing datasets individually we found methylation data provided the best predictions with an accuracy of 0.63 (95%CI = [0.54-0.71]), followed by RNA, 0.61 (95%CI = [0.52-0.69]), SNP, 0.59 (95%CI = [0.51-0.68]) and proteomics, 0.58 (95%CI = [0.51-0.67]). After integration of the four datasets, predictions were dramatically improved with a resulting accuracy of 0.95 (95% CI = [0.89-0.98]).</p><p><strong>Conclusion: </strong>The integration of data from multiple platforms is a powerful approach to explore biological systems and better characterise the biological signatures of AD. The results suggest that integrative methods can identify biomarker panels with improved predictive performance compared to individual platforms alone. Further validation in independent cohorts is required to validate and refine the results presented in this study.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"4 ","pages":"1390607"},"PeriodicalIF":3.9000,"publicationDate":"2024-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11219798/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2024.1390607","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Complex disorders, such as Alzheimer's disease (AD), result from the combined influence of multiple biological and environmental factors. The integration of high-throughput data from multiple omics platforms can provide system overviews, improving our understanding of complex biological processes underlying human disease. In this study, integrated data from four omics platforms were used to characterise biological signatures of AD.
Method: The study cohort consists of 455 participants (Control:148, Cases:307) from the Religious Orders Study and Memory and Aging Project (ROSMAP). Genotype (SNP), methylation (CpG), RNA and proteomics data were collected, quality-controlled and pre-processed (SNP = 130; CpG = 83; RNA = 91; Proteomics = 119). Using a diagnosis of Mild Cognitive Impairment (MCI)/AD combined as the target phenotype, we first used Partial Least Squares Regression as an unsupervised classification framework to assess the prediction capabilities for each omics dataset individually. We then used a variation of the sparse generalized canonical correlation analysis (sGCCA) to assess predictions of the combined datasets and identify multi-omics signatures characterising each group of participants.
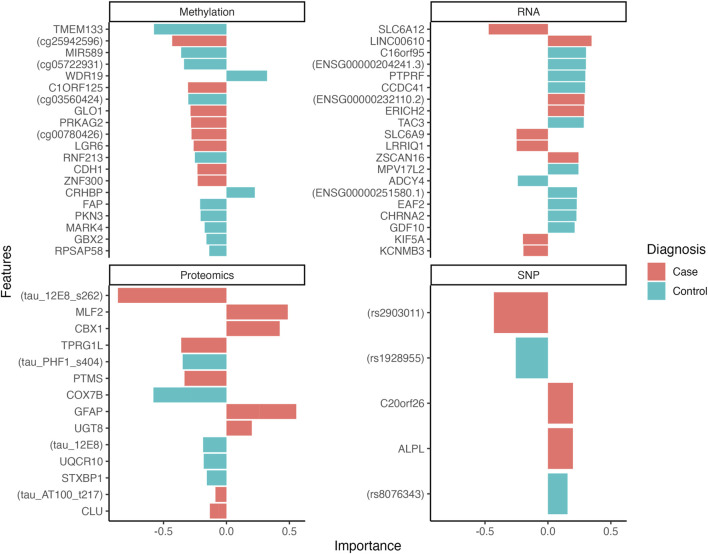
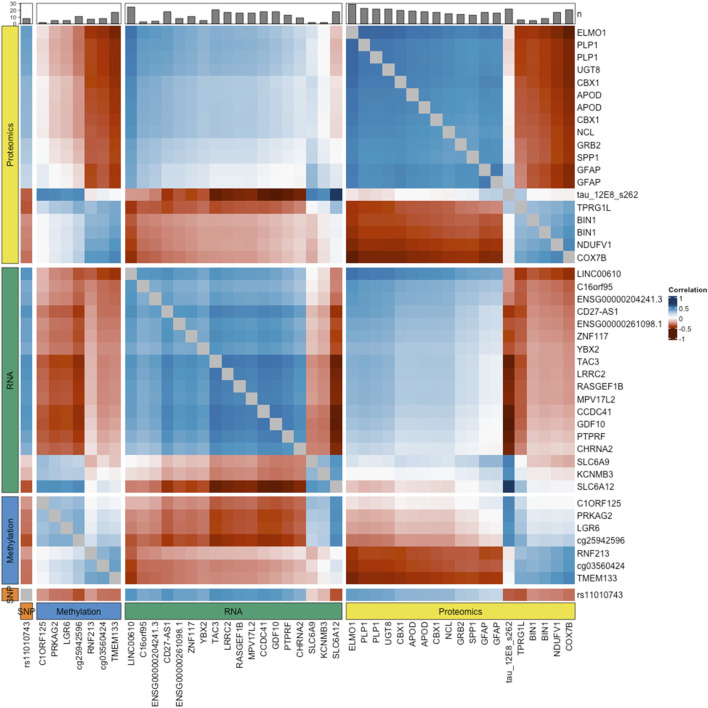
Results: Analysing datasets individually we found methylation data provided the best predictions with an accuracy of 0.63 (95%CI = [0.54-0.71]), followed by RNA, 0.61 (95%CI = [0.52-0.69]), SNP, 0.59 (95%CI = [0.51-0.68]) and proteomics, 0.58 (95%CI = [0.51-0.67]). After integration of the four datasets, predictions were dramatically improved with a resulting accuracy of 0.95 (95% CI = [0.89-0.98]).
Conclusion: The integration of data from multiple platforms is a powerful approach to explore biological systems and better characterise the biological signatures of AD. The results suggest that integrative methods can identify biomarker panels with improved predictive performance compared to individual platforms alone. Further validation in independent cohorts is required to validate and refine the results presented in this study.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
