Md. Shahadat Hossain, Md. Siddik Alom, Mohammad Salauddin Kader, Mohammed Akhter Hossain, Mohammad A. Halim
{"title":"Structure-Guided Antiviral Peptides Identification Targeting the HIV-1 Integrase","authors":"Md. Shahadat Hossain, Md. Siddik Alom, Mohammad Salauddin Kader, Mohammed Akhter Hossain, Mohammad A. Halim","doi":"10.1021/acsphyschemau.4c00006","DOIUrl":null,"url":null,"abstract":"HIV-1 integrase (IN), a major protein in the HIV life cycle responsible for integrating viral cDNA into the host DNA, represents a promising drug target. Small peptides have emerged as antiviral therapeutics for HIV because of their facile synthesis, highly selective nature, and fewer side effects. However, selecting the best candidates from a vast pool of peptides is a daunting task. In this study, multistep virtual screening was employed to identify potential peptides from a list of 280 HIV inhibitory peptides. Initially, 80 peptides were selected based on their minimum inhibitory concentrations (MIC). Then, molecular docking was performed to evaluate their binding scores compared to HIP000 and HIP00N which are experimentally validated HIV-1 integrase binding peptides that were used as a positive and negative control, respectively. The top-scoring docked complexes, namely, IN-HIP1113, IN-HIP1140, IN-HIP1142, IN-HIP678, IN-HIP776, and IN-HIP777, were subjected to initial 500 ns molecular dynamics (MD) simulations. Subsequently, HIP776, HIP777, and HIP1142 were selected for an in-depth mechanistic study of peptide interactions, with multiple simulations conducted for each complex spanning one microsecond. Independent simulations of the peptides, along with comparisons to the bound state, were performed to elucidate the conformational dynamics of the peptides. These peptides exhibit strong interactions with specific residues, as revealed by snapshot interaction analysis. Notably, LYS159, LYS156, VAL150, and GLU69 residues are prominently involved in these interactions. Additionally, residue-based binding free energy (BFE) calculations highlight the significance of HIS67, GLN148, GLN146, and SER147 residues within the binding pocket. Furthermore, the structure–activity relationship (SAR) analysis demonstrated that aromatic amino acids and the overall volume of peptides are the two major contributors to the docking scores. The best peptides will be validated experimentally by incorporating SAR properties, aiming to develop them as therapeutic agents and structural models for future peptide-based HIV-1 drug design, addressing the urgent need for effective HIV treatments.","PeriodicalId":29796,"journal":{"name":"ACS Physical Chemistry Au","volume":"14 1","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-07-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Physical Chemistry Au","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1021/acsphyschemau.4c00006","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
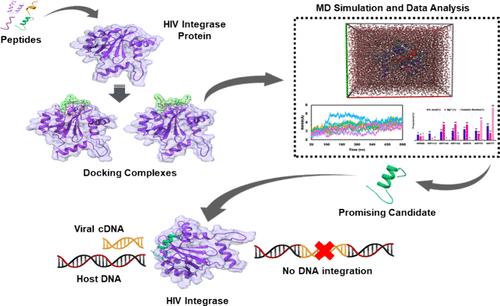
HIV-1 integrase (IN), a major protein in the HIV life cycle responsible for integrating viral cDNA into the host DNA, represents a promising drug target. Small peptides have emerged as antiviral therapeutics for HIV because of their facile synthesis, highly selective nature, and fewer side effects. However, selecting the best candidates from a vast pool of peptides is a daunting task. In this study, multistep virtual screening was employed to identify potential peptides from a list of 280 HIV inhibitory peptides. Initially, 80 peptides were selected based on their minimum inhibitory concentrations (MIC). Then, molecular docking was performed to evaluate their binding scores compared to HIP000 and HIP00N which are experimentally validated HIV-1 integrase binding peptides that were used as a positive and negative control, respectively. The top-scoring docked complexes, namely, IN-HIP1113, IN-HIP1140, IN-HIP1142, IN-HIP678, IN-HIP776, and IN-HIP777, were subjected to initial 500 ns molecular dynamics (MD) simulations. Subsequently, HIP776, HIP777, and HIP1142 were selected for an in-depth mechanistic study of peptide interactions, with multiple simulations conducted for each complex spanning one microsecond. Independent simulations of the peptides, along with comparisons to the bound state, were performed to elucidate the conformational dynamics of the peptides. These peptides exhibit strong interactions with specific residues, as revealed by snapshot interaction analysis. Notably, LYS159, LYS156, VAL150, and GLU69 residues are prominently involved in these interactions. Additionally, residue-based binding free energy (BFE) calculations highlight the significance of HIS67, GLN148, GLN146, and SER147 residues within the binding pocket. Furthermore, the structure–activity relationship (SAR) analysis demonstrated that aromatic amino acids and the overall volume of peptides are the two major contributors to the docking scores. The best peptides will be validated experimentally by incorporating SAR properties, aiming to develop them as therapeutic agents and structural models for future peptide-based HIV-1 drug design, addressing the urgent need for effective HIV treatments.
期刊介绍:
ACS Physical Chemistry Au is an open access journal which publishes original fundamental and applied research on all aspects of physical chemistry. The journal publishes new and original experimental computational and theoretical research of interest to physical chemists biophysical chemists chemical physicists physicists material scientists and engineers. An essential criterion for acceptance is that the manuscript provides new physical insight or develops new tools and methods of general interest. Some major topical areas include:Molecules Clusters and Aerosols; Biophysics Biomaterials Liquids and Soft Matter; Energy Materials and Catalysis


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
