{"title":"DeltaGzip: Computing Biopolymer-Ligand Binding Affinity via Kolmogorov Complexity and Lossless Compression.","authors":"Tao Liu, Lena Simine","doi":"10.1021/acs.jcim.4c00461","DOIUrl":null,"url":null,"abstract":"<p><p>The design of biosequences for biosensing and therapeutics is a challenging multistep search and optimization task. In principle, computational modeling may speed up the design process by virtual screening of sequences based on their binding affinities to target molecules. However, in practice, existing machine-learned models trained to predict binding affinities lack the flexibility with respect to reaction conditions, and molecular dynamics simulations that can incorporate reaction conditions suffer from high computational costs. Here, we describe a computational approach called DeltaGzip that evaluates the free energy of binding in biopolymer-ligand complexes from ultrashort equilibrium molecular dynamics simulations. The entropy of binding is evaluated using the Kolmogorov complexity definition of entropy and approximated using a lossless compression algorithm, Gzip. We benchmark the method on a well-studied data set of protein-ligand complexes comparing the predictions of DeltaGzip to the free energies of binding obtained using Jarzynski equality and experimental measurements.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5617-5623"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00461","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/9 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
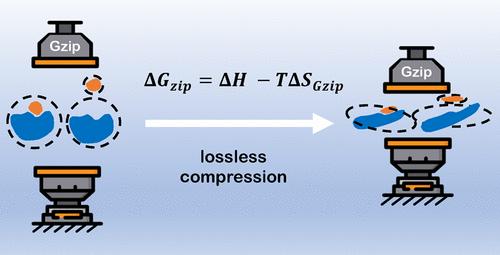
The design of biosequences for biosensing and therapeutics is a challenging multistep search and optimization task. In principle, computational modeling may speed up the design process by virtual screening of sequences based on their binding affinities to target molecules. However, in practice, existing machine-learned models trained to predict binding affinities lack the flexibility with respect to reaction conditions, and molecular dynamics simulations that can incorporate reaction conditions suffer from high computational costs. Here, we describe a computational approach called DeltaGzip that evaluates the free energy of binding in biopolymer-ligand complexes from ultrashort equilibrium molecular dynamics simulations. The entropy of binding is evaluated using the Kolmogorov complexity definition of entropy and approximated using a lossless compression algorithm, Gzip. We benchmark the method on a well-studied data set of protein-ligand complexes comparing the predictions of DeltaGzip to the free energies of binding obtained using Jarzynski equality and experimental measurements.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
