Nicolas Oldani, Victor M Freixas, Dianelys Ondarse-Alvarez, Sahar Sharifzadeh, Tammie Gibson, Sergei Tretiak, Sebastian Fernandez-Alberti
{"title":"Electronic Couplings versus Thermal Fluctuations in the Internal Conversion of Perylene Diimides: The Battle to Localize the Exciton.","authors":"Nicolas Oldani, Victor M Freixas, Dianelys Ondarse-Alvarez, Sahar Sharifzadeh, Tammie Gibson, Sergei Tretiak, Sebastian Fernandez-Alberti","doi":"10.1021/acs.jctc.4c00486","DOIUrl":null,"url":null,"abstract":"<p><p>Energy transfer processes among units of light-harvesting homo-oligomers impact the efficiency of these materials as components in organic optoelectronic devices such as solar cells. Perylene diimide (PDI), a prototypical dye, features exceptional light absorption and highly tunable optical and electronic properties. These properties can be modulated by varying the number of PDI units and linkers between them. Herein, atomistic nonadiabatic excited state molecular dynamics is used to explore the energy transfer during the internal conversion of acetylene and diacetylene bridged dimeric and trimeric PDIs. Our simulations reveal a significant impact of the bridge type on the transient exciton localization/delocalization between units of PDI dimers. After electronic relaxation, larger exciton delocalization occurs in the PDI dimer connected by the diacetylene bridge with respect to the one connected by the shorter acetylene bridge. These changes can be rationalized by the Frenkel exciton model. We outline a technique for deriving parameters for this model using inputs provided by nonadiabatic dynamics simulations. Frenkel exciton description reveals an interplay between the relative strengths of the diagonal and off-diagonal disorders. Moreover, atomistic simulations and the Frenkel exciton model of the PDI trimer systems corroborate in detail the localization properties of the exciton on the molecular units during the internal conversion to the lowest-energy excited state when the units become effectively decoupled. Overall, atomistic nonadiabatic simulations in combination with the Frenkel exciton model can serve as a predictive framework for analyzing and predicting desired exciton traps in PDI-based oligomers designed for organic electronics and photonic devices.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"5820-5828"},"PeriodicalIF":5.5000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00486","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/10 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
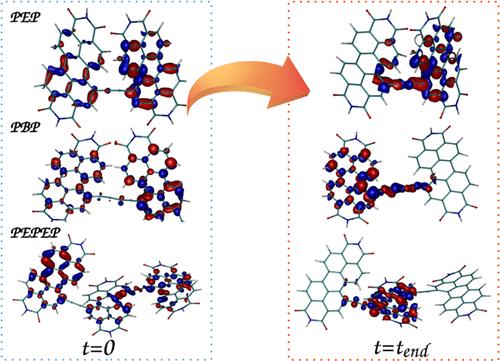
Energy transfer processes among units of light-harvesting homo-oligomers impact the efficiency of these materials as components in organic optoelectronic devices such as solar cells. Perylene diimide (PDI), a prototypical dye, features exceptional light absorption and highly tunable optical and electronic properties. These properties can be modulated by varying the number of PDI units and linkers between them. Herein, atomistic nonadiabatic excited state molecular dynamics is used to explore the energy transfer during the internal conversion of acetylene and diacetylene bridged dimeric and trimeric PDIs. Our simulations reveal a significant impact of the bridge type on the transient exciton localization/delocalization between units of PDI dimers. After electronic relaxation, larger exciton delocalization occurs in the PDI dimer connected by the diacetylene bridge with respect to the one connected by the shorter acetylene bridge. These changes can be rationalized by the Frenkel exciton model. We outline a technique for deriving parameters for this model using inputs provided by nonadiabatic dynamics simulations. Frenkel exciton description reveals an interplay between the relative strengths of the diagonal and off-diagonal disorders. Moreover, atomistic simulations and the Frenkel exciton model of the PDI trimer systems corroborate in detail the localization properties of the exciton on the molecular units during the internal conversion to the lowest-energy excited state when the units become effectively decoupled. Overall, atomistic nonadiabatic simulations in combination with the Frenkel exciton model can serve as a predictive framework for analyzing and predicting desired exciton traps in PDI-based oligomers designed for organic electronics and photonic devices.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
