Samiha Sharlin, Rodrigo A Lozano, Tyler R Josephson
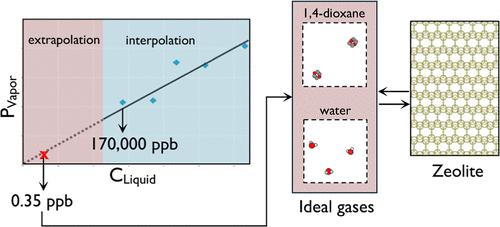
{"title":"Monte Carlo Simulations of Water Pollutant Adsorption at Parts-per-Billion Concentration: A Study on 1,4-Dioxane.","authors":"Samiha Sharlin, Rodrigo A Lozano, Tyler R Josephson","doi":"10.1021/acs.jctc.4c00236","DOIUrl":null,"url":null,"abstract":"<p><p>1,4-dioxane, an emerging water pollutant with high production volumes, is a probable human carcinogen. The inadequacy of conventional treatment processes demonstrates the need for an effective remediation strategy. Crystalline nanoporous materials are cost-effective adsorbents due to their high capacity and selective separation in mixtures. This study explores the potential of all-silica zeolites for the separation of 1,4-dioxane from water. These zeolites are highly hydrophobic and can preferentially adsorb nonpolar molecules from mixtures. We investigated six zeolite frameworks (BEA, EUO, FER, IFR, MFI, and MOR) using Monte Carlo simulations in the Gibbs ensemble. The simulations indicate high selectivity by FER and EUO, especially at low pressures, which we attribute to pore sizes and shapes with a greater affinity to 1,4-dioxane. We also demonstrate a Monte Carlo simulation workflow using gauge cells to model the adsorption of an aqueous solution of 1,4-dioxane at a 0.35 ppb concentration. We quantify 1,4-dioxane and water coadsorption and observe selectivities ranging from 1.1 × 10<sup>5</sup> in MOR to 8.7 × 10<sup>6</sup> in FER. We also demonstrate that 1,4-dioxane is in the infinite dilution regime in the aqueous phase at this concentration. This simulation technique can be extended to model other emerging water contaminants such as perfluoroalkyl and polyfluoroalkyl substances (PFAS), chlorofluorocarbons, and others, which are also found in extremely low concentrations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"5854-5865"},"PeriodicalIF":5.5000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00236","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/10 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
1,4-dioxane, an emerging water pollutant with high production volumes, is a probable human carcinogen. The inadequacy of conventional treatment processes demonstrates the need for an effective remediation strategy. Crystalline nanoporous materials are cost-effective adsorbents due to their high capacity and selective separation in mixtures. This study explores the potential of all-silica zeolites for the separation of 1,4-dioxane from water. These zeolites are highly hydrophobic and can preferentially adsorb nonpolar molecules from mixtures. We investigated six zeolite frameworks (BEA, EUO, FER, IFR, MFI, and MOR) using Monte Carlo simulations in the Gibbs ensemble. The simulations indicate high selectivity by FER and EUO, especially at low pressures, which we attribute to pore sizes and shapes with a greater affinity to 1,4-dioxane. We also demonstrate a Monte Carlo simulation workflow using gauge cells to model the adsorption of an aqueous solution of 1,4-dioxane at a 0.35 ppb concentration. We quantify 1,4-dioxane and water coadsorption and observe selectivities ranging from 1.1 × 105 in MOR to 8.7 × 106 in FER. We also demonstrate that 1,4-dioxane is in the infinite dilution regime in the aqueous phase at this concentration. This simulation technique can be extended to model other emerging water contaminants such as perfluoroalkyl and polyfluoroalkyl substances (PFAS), chlorofluorocarbons, and others, which are also found in extremely low concentrations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
