Hongwei Liu, Wei Zhang, Yihao Zhang, Abraham Ayodeji Adegboro, Deborah Oluwatosin Fasoranti, Luohuan Dai, Zhouyang Pan, Hongyi Liu, Yi Xiong, Wang Li, Kang Peng, Siyi Wanggou, Xuejun Li
{"title":"Mime: A flexible machine-learning framework to construct and visualize models for clinical characteristics prediction and feature selection","authors":"Hongwei Liu, Wei Zhang, Yihao Zhang, Abraham Ayodeji Adegboro, Deborah Oluwatosin Fasoranti, Luohuan Dai, Zhouyang Pan, Hongyi Liu, Yi Xiong, Wang Li, Kang Peng, Siyi Wanggou, Xuejun Li","doi":"10.1016/j.csbj.2024.06.035","DOIUrl":null,"url":null,"abstract":"The widespread use of high-throughput sequencing technologies has revolutionized the understanding of biology and cancer heterogeneity. Recently, several machine-learning models based on transcriptional data have been developed to accurately predict patients’ outcome and clinical response. However, an open-source R package covering state-of-the-art machine-learning algorithms for user-friendly access has yet to be developed. Thus, we proposed a flexible computational framework to construct a machine learning-based integration model with elegant performance (Mime). Mime streamlines the process of developing predictive models with high accuracy, leveraging complex datasets to identify critical genes associated with prognosis. An in silico combined model based on de novo PIEZO1-associated signatures constructed by Mime demonstrated high accuracy in predicting the outcomes of patients compared with other published models. Furthermore, the PIEZO1-associated signatures could also precisely infer immunotherapy response by applying different algorithms in Mime. Finally, SDC1 selected from the PIEZO1-associated signatures demonstrated high potential as a glioma target. Taken together, our package provides a user-friendly solution for constructing machine learning-based integration models and will be greatly expanded to provide valuable insights into current fields. The Mime package is available on GitHub ().","PeriodicalId":10715,"journal":{"name":"Computational and structural biotechnology journal","volume":"14 1","pages":""},"PeriodicalIF":4.1000,"publicationDate":"2024-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and structural biotechnology journal","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.csbj.2024.06.035","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
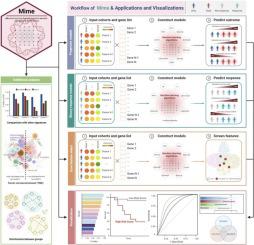
The widespread use of high-throughput sequencing technologies has revolutionized the understanding of biology and cancer heterogeneity. Recently, several machine-learning models based on transcriptional data have been developed to accurately predict patients’ outcome and clinical response. However, an open-source R package covering state-of-the-art machine-learning algorithms for user-friendly access has yet to be developed. Thus, we proposed a flexible computational framework to construct a machine learning-based integration model with elegant performance (Mime). Mime streamlines the process of developing predictive models with high accuracy, leveraging complex datasets to identify critical genes associated with prognosis. An in silico combined model based on de novo PIEZO1-associated signatures constructed by Mime demonstrated high accuracy in predicting the outcomes of patients compared with other published models. Furthermore, the PIEZO1-associated signatures could also precisely infer immunotherapy response by applying different algorithms in Mime. Finally, SDC1 selected from the PIEZO1-associated signatures demonstrated high potential as a glioma target. Taken together, our package provides a user-friendly solution for constructing machine learning-based integration models and will be greatly expanded to provide valuable insights into current fields. The Mime package is available on GitHub ().
期刊介绍:
Computational and Structural Biotechnology Journal (CSBJ) is an online gold open access journal publishing research articles and reviews after full peer review. All articles are published, without barriers to access, immediately upon acceptance. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together through the application of computational methods. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal. Specific areas of interest include, but are not limited to:
Structure and function of proteins, nucleic acids and other macromolecules
Structure and function of multi-component complexes
Protein folding, processing and degradation
Enzymology
Computational and structural studies of plant systems
Microbial Informatics
Genomics
Proteomics
Metabolomics
Algorithms and Hypothesis in Bioinformatics
Mathematical and Theoretical Biology
Computational Chemistry and Drug Discovery
Microscopy and Molecular Imaging
Nanotechnology
Systems and Synthetic Biology



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
