Jianshu Zhao, Jean Pierre Both, Luis M Rodriguez-R, Konstantinos T Konstantinidis
{"title":"GSearch: ultra-fast and scalable genome search by combining K-mer hashing with hierarchical navigable small world graphs.","authors":"Jianshu Zhao, Jean Pierre Both, Luis M Rodriguez-R, Konstantinos T Konstantinidis","doi":"10.1093/nar/gkae609","DOIUrl":null,"url":null,"abstract":"<p><p>Genome search and/or classification typically involves finding the best-match database (reference) genomes and has become increasingly challenging due to the growing number of available database genomes and the fact that traditional methods do not scale well with large databases. By combining k-mer hashing-based probabilistic data structures (i.e. ProbMinHash, SuperMinHash, Densified MinHash and SetSketch) to estimate genomic distance, with a graph based nearest neighbor search algorithm (Hierarchical Navigable Small World Graphs, or HNSW), we created a new data structure and developed an associated computer program, GSearch, that is orders of magnitude faster than alternative tools while maintaining high accuracy and low memory usage. For example, GSearch can search 8000 query genomes against all available microbial or viral genomes for their best matches (n = ∼318 000 or ∼3 000 000, respectively) within a few minutes on a personal laptop, using ∼6 GB of memory (2.5 GB via SetSketch). Notably, GSearch has an O(log(N)) time complexity and will scale well with billions of genomes based on a database splitting strategy. Further, GSearch implements a three-step search strategy depending on the degree of novelty of the query genomes to maximize specificity and sensitivity. Therefore, GSearch solves a major bottleneck of microbiome studies that require genome search and/or classification.</p>","PeriodicalId":19471,"journal":{"name":"Nucleic Acids Research","volume":" ","pages":"e74"},"PeriodicalIF":13.1000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11381346/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nucleic Acids Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/nar/gkae609","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
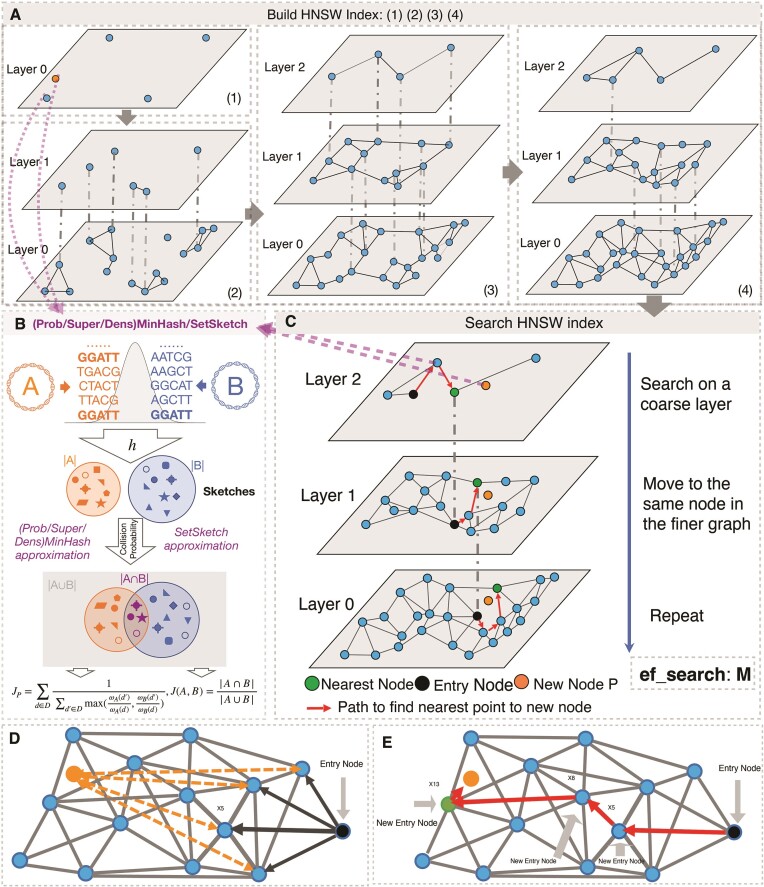
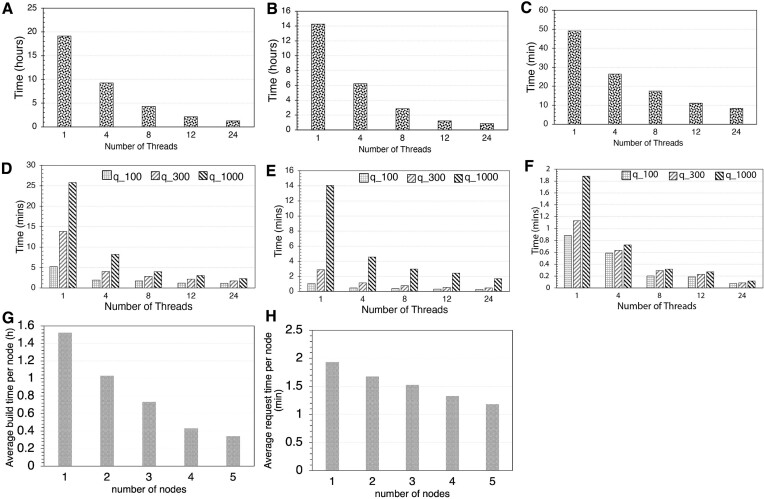
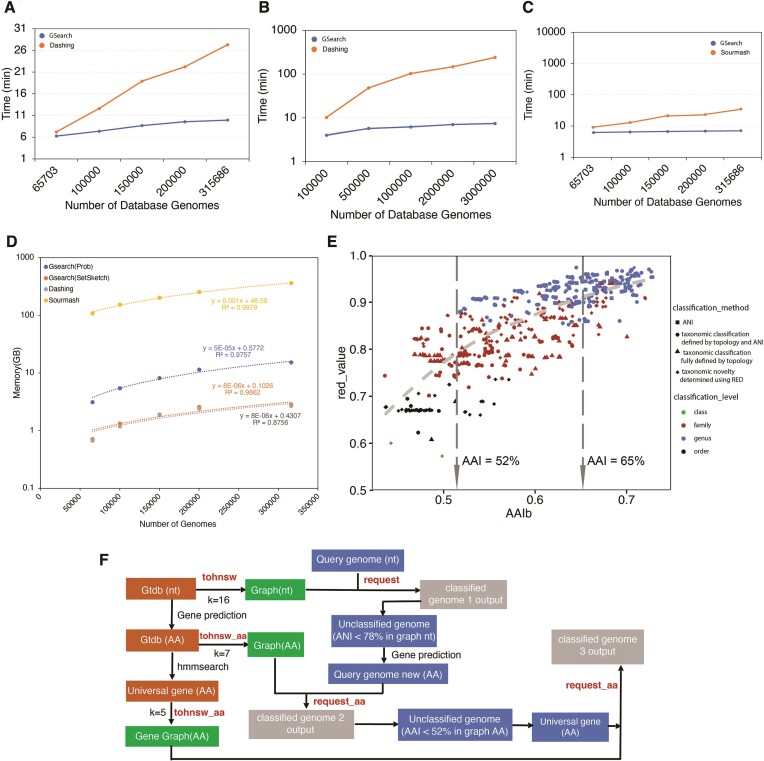
Genome search and/or classification typically involves finding the best-match database (reference) genomes and has become increasingly challenging due to the growing number of available database genomes and the fact that traditional methods do not scale well with large databases. By combining k-mer hashing-based probabilistic data structures (i.e. ProbMinHash, SuperMinHash, Densified MinHash and SetSketch) to estimate genomic distance, with a graph based nearest neighbor search algorithm (Hierarchical Navigable Small World Graphs, or HNSW), we created a new data structure and developed an associated computer program, GSearch, that is orders of magnitude faster than alternative tools while maintaining high accuracy and low memory usage. For example, GSearch can search 8000 query genomes against all available microbial or viral genomes for their best matches (n = ∼318 000 or ∼3 000 000, respectively) within a few minutes on a personal laptop, using ∼6 GB of memory (2.5 GB via SetSketch). Notably, GSearch has an O(log(N)) time complexity and will scale well with billions of genomes based on a database splitting strategy. Further, GSearch implements a three-step search strategy depending on the degree of novelty of the query genomes to maximize specificity and sensitivity. Therefore, GSearch solves a major bottleneck of microbiome studies that require genome search and/or classification.
期刊介绍:
Nucleic Acids Research (NAR) is a scientific journal that publishes research on various aspects of nucleic acids and proteins involved in nucleic acid metabolism and interactions. It covers areas such as chemistry and synthetic biology, computational biology, gene regulation, chromatin and epigenetics, genome integrity, repair and replication, genomics, molecular biology, nucleic acid enzymes, RNA, and structural biology. The journal also includes a Survey and Summary section for brief reviews. Additionally, each year, the first issue is dedicated to biological databases, and an issue in July focuses on web-based software resources for the biological community. Nucleic Acids Research is indexed by several services including Abstracts on Hygiene and Communicable Diseases, Animal Breeding Abstracts, Agricultural Engineering Abstracts, Agbiotech News and Information, BIOSIS Previews, CAB Abstracts, and EMBASE.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
