{"title":"Universal neural network potentials as descriptors: towards scalable chemical property prediction using quantum and classical computers","authors":"Tomoya Shiota, Kenji Ishihara and Wataru Mizukami","doi":"10.1039/D4DD00098F","DOIUrl":null,"url":null,"abstract":"<p >Accurate prediction of diverse chemical properties is crucial for advancing molecular design and materials discovery. Here we present a versatile approach that uses the intermediate information of a universal neural network potential as a general-purpose descriptor for chemical property prediction. Our method is based on the insight that by training a sophisticated neural network architecture for universal force fields, it learns transferable representations of atomic environments. We show that transfer learning with graph neural network potentials such as M3GNet and MACE achieves accuracy comparable to state-of-the-art methods for predicting the NMR chemical shifts by using quantum machine learning as well as a standard classical regression model, despite the compactness of its descriptors. In particular, the MACE descriptor demonstrates the highest accuracy to date on the <small><sup>13</sup></small>C NMR chemical shift benchmarks for drug molecules. This work provides an efficient way to accurately predict properties, potentially accelerating the discovery of new molecules and materials.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 9","pages":" 1714-1728"},"PeriodicalIF":6.2000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00098f?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00098f","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
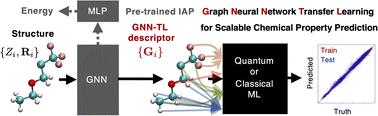
Abstract
Accurate prediction of diverse chemical properties is crucial for advancing molecular design and materials discovery. Here we present a versatile approach that uses the intermediate information of a universal neural network potential as a general-purpose descriptor for chemical property prediction. Our method is based on the insight that by training a sophisticated neural network architecture for universal force fields, it learns transferable representations of atomic environments. We show that transfer learning with graph neural network potentials such as M3GNet and MACE achieves accuracy comparable to state-of-the-art methods for predicting the NMR chemical shifts by using quantum machine learning as well as a standard classical regression model, despite the compactness of its descriptors. In particular, the MACE descriptor demonstrates the highest accuracy to date on the 13C NMR chemical shift benchmarks for drug molecules. This work provides an efficient way to accurately predict properties, potentially accelerating the discovery of new molecules and materials.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
