Madalynn L Erb, Kayla Sipple, Nathan Levine, Xi Chen, Darren J Moore
{"title":"Adult-onset deletion of ATP13A2 in mice induces progressive nigrostriatal pathway dopaminergic degeneration and lysosomal abnormalities.","authors":"Madalynn L Erb, Kayla Sipple, Nathan Levine, Xi Chen, Darren J Moore","doi":"10.1038/s41531-024-00748-5","DOIUrl":null,"url":null,"abstract":"<p><p>Although most cases of Parkinson's disease (PD) are sporadic, mutations in over 20 genes are known to cause heritable forms of the disease. Recessive loss-of-function mutations in ATP13A2, a lysosomal transmembrane P5<sub>B</sub>-type ATPase and polyamine exporter, can cause early-onset familial PD. Familial ATP13A2 mutations are also linked to related neurodegenerative diseases, including Kufor-Rakeb syndrome, hereditary spastic paraplegias, neuronal ceroid lipofuscinosis, and amyotrophic lateral sclerosis. Despite the severe effects of ATP13A2 mutations in humans, ATP13A2 knockout (KO) mice fail to exhibit neurodegeneration even at advanced ages, making it challenging to study the neuropathological effects of ATP13A2 loss in vivo. Germline deletion of ATP13A2 in rodents may trigger the upregulation of compensatory pathways during embryonic development that mask the full neurotoxic effects of ATP13A2 loss in the brain. To explore this idea, we selectively deleted ATP13A2 in the adult mouse brain by the unilateral delivery of an AAV-Cre vector into the substantia nigra of young adult mice carrying conditional loxP-flanked ATP13A2 KO alleles. We observe a progressive loss of striatal dopaminergic nerve terminals at 3 and 10 months after AAV-Cre delivery. Cre-injected mice also exhibit robust dopaminergic neuronal degeneration in the substantia nigra at 10 months. Adult-onset ATP13A2 KO also recreates many of the phenotypes observed in aged germline ATP13A2 KO mice, including lysosomal abnormalities, p62-positive inclusions, and neuroinflammation. Our study demonstrates that the adult-onset homozygous deletion of ATP13A2 in the nigrostriatal pathway produces robust and progressive dopaminergic neurodegeneration that serves as a useful in vivo model of ATP13A2-related neurodegenerative diseases.</p>","PeriodicalId":19706,"journal":{"name":"NPJ Parkinson's Disease","volume":"10 1","pages":"133"},"PeriodicalIF":8.2000,"publicationDate":"2024-07-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11271504/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Parkinson's Disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41531-024-00748-5","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
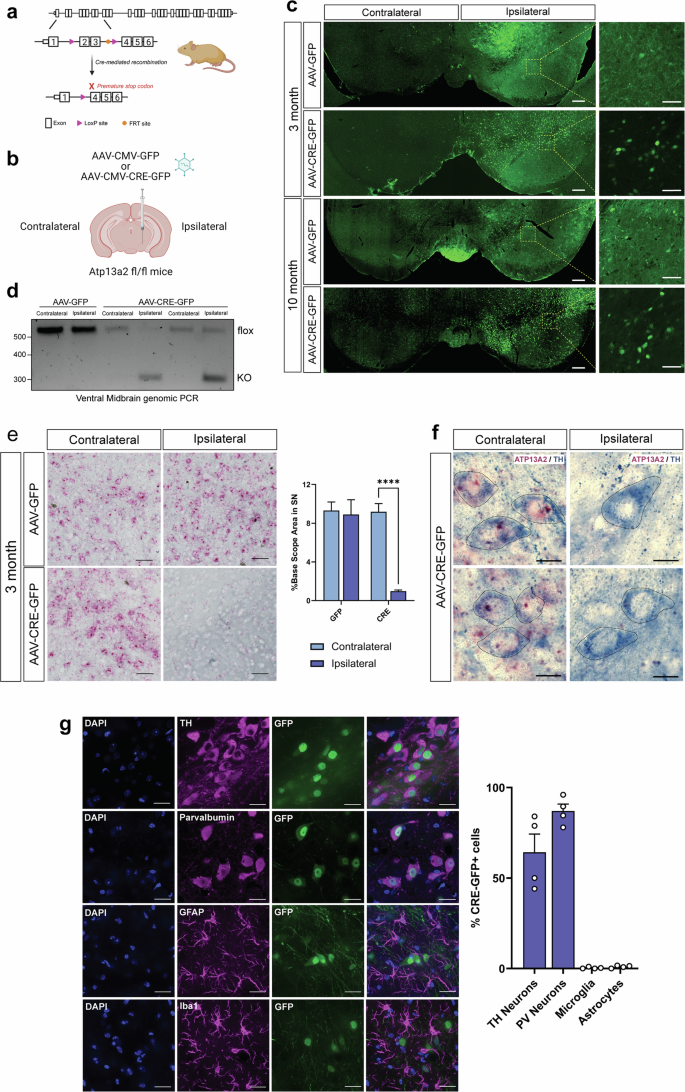
Although most cases of Parkinson's disease (PD) are sporadic, mutations in over 20 genes are known to cause heritable forms of the disease. Recessive loss-of-function mutations in ATP13A2, a lysosomal transmembrane P5B-type ATPase and polyamine exporter, can cause early-onset familial PD. Familial ATP13A2 mutations are also linked to related neurodegenerative diseases, including Kufor-Rakeb syndrome, hereditary spastic paraplegias, neuronal ceroid lipofuscinosis, and amyotrophic lateral sclerosis. Despite the severe effects of ATP13A2 mutations in humans, ATP13A2 knockout (KO) mice fail to exhibit neurodegeneration even at advanced ages, making it challenging to study the neuropathological effects of ATP13A2 loss in vivo. Germline deletion of ATP13A2 in rodents may trigger the upregulation of compensatory pathways during embryonic development that mask the full neurotoxic effects of ATP13A2 loss in the brain. To explore this idea, we selectively deleted ATP13A2 in the adult mouse brain by the unilateral delivery of an AAV-Cre vector into the substantia nigra of young adult mice carrying conditional loxP-flanked ATP13A2 KO alleles. We observe a progressive loss of striatal dopaminergic nerve terminals at 3 and 10 months after AAV-Cre delivery. Cre-injected mice also exhibit robust dopaminergic neuronal degeneration in the substantia nigra at 10 months. Adult-onset ATP13A2 KO also recreates many of the phenotypes observed in aged germline ATP13A2 KO mice, including lysosomal abnormalities, p62-positive inclusions, and neuroinflammation. Our study demonstrates that the adult-onset homozygous deletion of ATP13A2 in the nigrostriatal pathway produces robust and progressive dopaminergic neurodegeneration that serves as a useful in vivo model of ATP13A2-related neurodegenerative diseases.
期刊介绍:
npj Parkinson's Disease is a comprehensive open access journal that covers a wide range of research areas related to Parkinson's disease. It publishes original studies in basic science, translational research, and clinical investigations. The journal is dedicated to advancing our understanding of Parkinson's disease by exploring various aspects such as anatomy, etiology, genetics, cellular and molecular physiology, neurophysiology, epidemiology, and therapeutic development. By providing free and immediate access to the scientific and Parkinson's disease community, npj Parkinson's Disease promotes collaboration and knowledge sharing among researchers and healthcare professionals.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
