Mar Costa-Roger, Laura Blasco-Pérez, Lorene Gerin, Marta Codina-Solà, Jordi Leno-Colorado, Marta Gómez-García De la Banda, Rocio Garcia-Uzquiano, Pascale Saugier-Veber, Séverine Drunat, Susana Quijano-Roy, Eduardo F Tizzano
{"title":"Complex <i>SMN</i> Hybrids Detected in a Cohort of 31 Patients With Spinal Muscular Atrophy.","authors":"Mar Costa-Roger, Laura Blasco-Pérez, Lorene Gerin, Marta Codina-Solà, Jordi Leno-Colorado, Marta Gómez-García De la Banda, Rocio Garcia-Uzquiano, Pascale Saugier-Veber, Séverine Drunat, Susana Quijano-Roy, Eduardo F Tizzano","doi":"10.1212/NXG.0000000000200175","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Spinal muscular atrophy (SMA) is a recessive neuromuscular disorder caused by the loss or presence of point pathogenic variants in the <i>SMN1</i> gene. The main positive modifier of the SMA phenotype is the number of copies of the <i>SMN2</i> gene, a paralog of <i>SMN1</i>, which only produces around 10%-15% of functional SMN protein. The <i>SMN2</i> copy number is inversely correlated with phenotype severity; however, discrepancies between the SMA type and the <i>SMN2</i> copy number have been reported. The presence of <i>SMN2-SMN1</i> hybrids has been proposed as a possible modifier of SMA disease.</p><p><strong>Methods: </strong>We studied 31 patients with SMA, followed at a single center and molecularly diagnosed by Multiplex Ligand-Dependent Probe Amplification (MLPA), with a specific next-generation sequencing protocol to investigate their <i>SMN2</i> genes in depth. Hybrid characterization also included bioinformatics haplotype phasing and specific PCRs to resolve each <i>SMN2-SMN1</i> hybrid structure.</p><p><strong>Results: </strong>We detected <i>SMN2-SMN1</i> hybrid genes in 45.2% of the patients (14/31), the highest rate reported to date. This represents a total of 25 hybrid alleles, with 9 different structures, of which only 4 are detectable by MLPA. Of particular interest were 2 patients who presented 4 <i>SMN2-SMN1</i> hybrid copies each and no pure <i>SMN2</i> copies, an event reported here for the first time. No clear trend between the presence of hybrids and a milder phenotype was observed, although 5 of the patients with hybrid copies showed a better-than-expected phenotype. The higher hybrid detection rate in our cohort may be due to both the methodology applied, which allows an in-depth characterization of the <i>SMN</i> genes and the ethnicity of the patients, mainly of African origin.</p><p><strong>Discussion: </strong>Although hybrid genes have been proposed to be beneficial for patients with SMA, our work revealed great complexity and variability between hybrid structures; therefore, each hybrid structure should be studied independently to determine its contribution to the SMA phenotype. Large-scale studies are needed to gain a better understanding of the function and implications of <i>SMN2-SMN1</i> hybrid copies, improving genotype-phenotype correlations and prediction of the evolution of patients with SMA.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"10 4","pages":"e200175"},"PeriodicalIF":3.7000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11259531/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200175","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objectives: Spinal muscular atrophy (SMA) is a recessive neuromuscular disorder caused by the loss or presence of point pathogenic variants in the SMN1 gene. The main positive modifier of the SMA phenotype is the number of copies of the SMN2 gene, a paralog of SMN1, which only produces around 10%-15% of functional SMN protein. The SMN2 copy number is inversely correlated with phenotype severity; however, discrepancies between the SMA type and the SMN2 copy number have been reported. The presence of SMN2-SMN1 hybrids has been proposed as a possible modifier of SMA disease.
Methods: We studied 31 patients with SMA, followed at a single center and molecularly diagnosed by Multiplex Ligand-Dependent Probe Amplification (MLPA), with a specific next-generation sequencing protocol to investigate their SMN2 genes in depth. Hybrid characterization also included bioinformatics haplotype phasing and specific PCRs to resolve each SMN2-SMN1 hybrid structure.
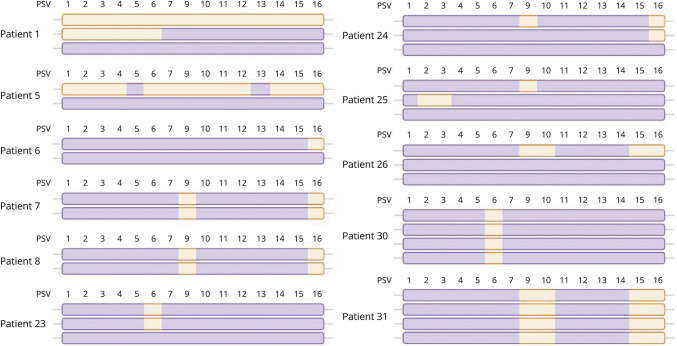
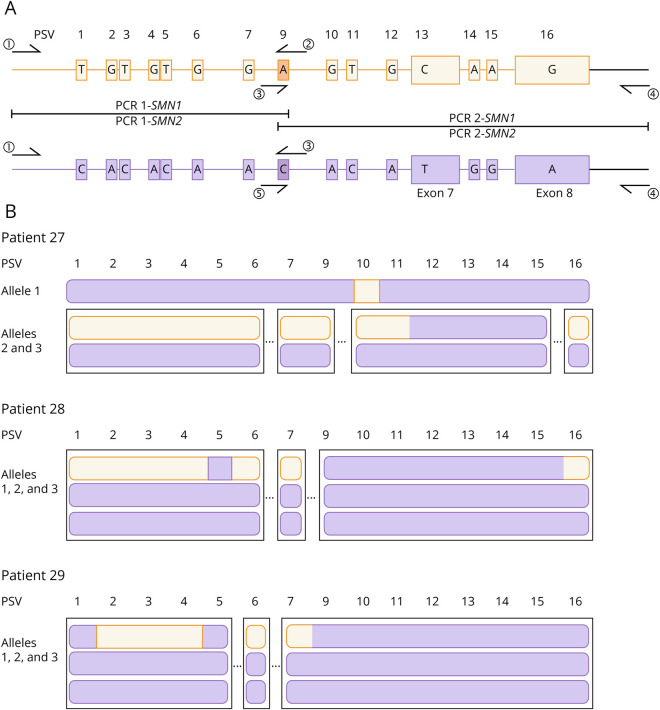
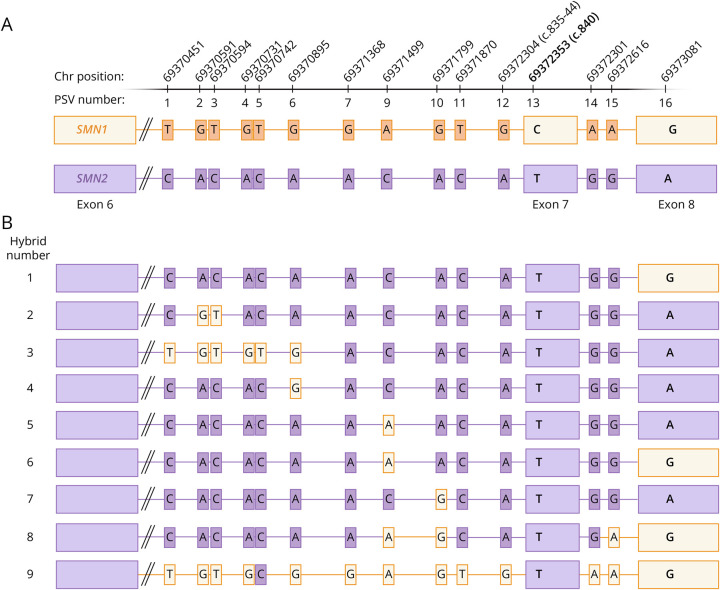
Results: We detected SMN2-SMN1 hybrid genes in 45.2% of the patients (14/31), the highest rate reported to date. This represents a total of 25 hybrid alleles, with 9 different structures, of which only 4 are detectable by MLPA. Of particular interest were 2 patients who presented 4 SMN2-SMN1 hybrid copies each and no pure SMN2 copies, an event reported here for the first time. No clear trend between the presence of hybrids and a milder phenotype was observed, although 5 of the patients with hybrid copies showed a better-than-expected phenotype. The higher hybrid detection rate in our cohort may be due to both the methodology applied, which allows an in-depth characterization of the SMN genes and the ethnicity of the patients, mainly of African origin.
Discussion: Although hybrid genes have been proposed to be beneficial for patients with SMA, our work revealed great complexity and variability between hybrid structures; therefore, each hybrid structure should be studied independently to determine its contribution to the SMA phenotype. Large-scale studies are needed to gain a better understanding of the function and implications of SMN2-SMN1 hybrid copies, improving genotype-phenotype correlations and prediction of the evolution of patients with SMA.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
