{"title":"The lethal homozygous variant in the ATP1A2 gene is associated with FARIMPD syndrome phenotypes in newborns.","authors":"Behzad Haj Mohammad Hassani, Kianoosh Malekzadeh","doi":"10.1007/s10048-024-00775-7","DOIUrl":null,"url":null,"abstract":"<p><p>FARIMPD (Fetal akinesia, respiratory insufficiency, microcephaly, polymicrogyria, and dysmorphic facies) syndrome is a severe condition caused by ATP1A2 gene variants. The syndrome's novelty and rarity have limited its clinical and molecular knowledge. This research tries to provide new insight by investigating the cause of the early deaths due to FARIMPD syndrome in a particular family and reviewing previous studies. DNA and RNA were extracted from the blood samples of newborns and their parents, followed by whole exome sequencing and segregation analysis. A pathogenic homozygous nonsense variant (c.1234C > T: p.Arg412*) in the ATP1A2 gene was found in newborns. This variant is reported as homozygous for the first time. The migraine symptoms were the result of the heterozygous state of this particular variant, which supported the dominant inheritance pattern of this disease. Real-time PCR was used to analyze ATP1A2 gene expression in the newborns compared to parents and control subjects. The expression analysis also showed significant mRNA degradation in the newborns compared to heterozygous and healthy individuals, due to Nonsense-mediated mRNA Decay phenomena. Our study describes an ATP1A2 nonsense variant (c.1234C > T) that appears compatible with infant survival in the heterozygous and compound heterozygous states but is lethal in the homozygous state.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":" ","pages":"417-424"},"PeriodicalIF":1.2000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-024-00775-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/24 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
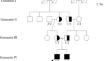
FARIMPD (Fetal akinesia, respiratory insufficiency, microcephaly, polymicrogyria, and dysmorphic facies) syndrome is a severe condition caused by ATP1A2 gene variants. The syndrome's novelty and rarity have limited its clinical and molecular knowledge. This research tries to provide new insight by investigating the cause of the early deaths due to FARIMPD syndrome in a particular family and reviewing previous studies. DNA and RNA were extracted from the blood samples of newborns and their parents, followed by whole exome sequencing and segregation analysis. A pathogenic homozygous nonsense variant (c.1234C > T: p.Arg412*) in the ATP1A2 gene was found in newborns. This variant is reported as homozygous for the first time. The migraine symptoms were the result of the heterozygous state of this particular variant, which supported the dominant inheritance pattern of this disease. Real-time PCR was used to analyze ATP1A2 gene expression in the newborns compared to parents and control subjects. The expression analysis also showed significant mRNA degradation in the newborns compared to heterozygous and healthy individuals, due to Nonsense-mediated mRNA Decay phenomena. Our study describes an ATP1A2 nonsense variant (c.1234C > T) that appears compatible with infant survival in the heterozygous and compound heterozygous states but is lethal in the homozygous state.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
