Florentin Lukas Holzem , Neil Parrott , Jeannine Petrig Schaffland , Martin Brandl , Annette Bauer-Brandl , Cordula Stillhart
{"title":"Oral Absorption from Surfactant-Based Drug Formulations: The Impact of Molecularly Dissolved Drug on Bioavailability","authors":"Florentin Lukas Holzem , Neil Parrott , Jeannine Petrig Schaffland , Martin Brandl , Annette Bauer-Brandl , Cordula Stillhart","doi":"10.1016/j.xphs.2024.07.017","DOIUrl":null,"url":null,"abstract":"<div><div>Enabling drug formulations are often required to ensure sufficient absorption after oral administration of poorly soluble drugs. While these formulations typically increase the apparent solubility of the drug, it is widely acknowledged that only molecularly dissolved, i.e., free fraction of the drug, is prone for direct absorption, while colloid-associated drug does not permeate to the same extent.</div><div>In the present study, we aimed at comparing the effect of molecularly and apparently (i.e., the sum of molecularly and colloid-associated drug) dissolved drug concentrations on the oral absorption of a poorly water-soluble drug compound, Alectinib. Mixtures of Alectinib and respectively 50 %, 25 %, 12.5 %, and 3 % sodium lauryl sulfate (SLS) relative to the dose were prepared and small-scale dissolution tests were performed under simulated fed and fasted state conditions. Both the molecularly and apparently dissolved drug concentrations were assessed in parallel using microdialysis and centrifugation/filtration sampling, respectively. The data served as the basis for an in vitro-in vivo correlation (IVIVC) and as input for a GastroPlus<sup>TM</sup> physiologically-based biopharmaceutics model (PBBM).</div><div>It was shown that with increasing the content of SLS the apparently dissolved drug in FeSSIF and FaSSIF increased to a linear extent and thus, the predicted in vivo performance of the 50 % SLS formulation, based on apparently dissolved drug, would outperform all other formulations. Against common expectation, however, the free (molecularly dissolved) drug concentrations were found to vary with SLS concentrations as well, yet to a minor extent. A systematic comparison of solubilized and free drug dissolution patterns at different SLS contents of the formulations and prandial states allowed for interesting insights into the complex dissolution-/supersaturation-, micellization-, and precipitation-behavior of the formulations. When comparing the in vitro datasets with human pharmacokinetic data from a bioequivalence study, it was shown that the use of molecularly dissolved drug resulted in an improved IVIVC.</div><div>By incorporating the in vitro dissolution datasets into the GastroPlus<sup>TM</sup> PBBM, the apparently dissolved drug concentrations resulted in both, a remarkable overprediction of plasma concentrations as well as a misprediction of the influence of SLS on systemic exposure. In contrast, by using the molecularly dissolved drug (i.e., free fraction) as the model input, the predicted plasma concentration-time profiles were in excellent agreement with observed data for all formulations under both fed and fasted conditions.</div><div>By combining an advanced in vitro assessment with PBBM, the present study confirmed that only the molecularly dissolved drug, and not the colloid-associated drug, is available for direct absorption.</div></div>","PeriodicalId":16741,"journal":{"name":"Journal of pharmaceutical sciences","volume":"113 10","pages":"Pages 3054-3064"},"PeriodicalIF":3.7000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of pharmaceutical sciences","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022354924002636","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Enabling drug formulations are often required to ensure sufficient absorption after oral administration of poorly soluble drugs. While these formulations typically increase the apparent solubility of the drug, it is widely acknowledged that only molecularly dissolved, i.e., free fraction of the drug, is prone for direct absorption, while colloid-associated drug does not permeate to the same extent.
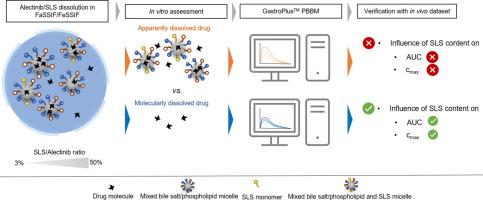
In the present study, we aimed at comparing the effect of molecularly and apparently (i.e., the sum of molecularly and colloid-associated drug) dissolved drug concentrations on the oral absorption of a poorly water-soluble drug compound, Alectinib. Mixtures of Alectinib and respectively 50 %, 25 %, 12.5 %, and 3 % sodium lauryl sulfate (SLS) relative to the dose were prepared and small-scale dissolution tests were performed under simulated fed and fasted state conditions. Both the molecularly and apparently dissolved drug concentrations were assessed in parallel using microdialysis and centrifugation/filtration sampling, respectively. The data served as the basis for an in vitro-in vivo correlation (IVIVC) and as input for a GastroPlusTM physiologically-based biopharmaceutics model (PBBM).
It was shown that with increasing the content of SLS the apparently dissolved drug in FeSSIF and FaSSIF increased to a linear extent and thus, the predicted in vivo performance of the 50 % SLS formulation, based on apparently dissolved drug, would outperform all other formulations. Against common expectation, however, the free (molecularly dissolved) drug concentrations were found to vary with SLS concentrations as well, yet to a minor extent. A systematic comparison of solubilized and free drug dissolution patterns at different SLS contents of the formulations and prandial states allowed for interesting insights into the complex dissolution-/supersaturation-, micellization-, and precipitation-behavior of the formulations. When comparing the in vitro datasets with human pharmacokinetic data from a bioequivalence study, it was shown that the use of molecularly dissolved drug resulted in an improved IVIVC.
By incorporating the in vitro dissolution datasets into the GastroPlusTM PBBM, the apparently dissolved drug concentrations resulted in both, a remarkable overprediction of plasma concentrations as well as a misprediction of the influence of SLS on systemic exposure. In contrast, by using the molecularly dissolved drug (i.e., free fraction) as the model input, the predicted plasma concentration-time profiles were in excellent agreement with observed data for all formulations under both fed and fasted conditions.
By combining an advanced in vitro assessment with PBBM, the present study confirmed that only the molecularly dissolved drug, and not the colloid-associated drug, is available for direct absorption.
期刊介绍:
The Journal of Pharmaceutical Sciences will publish original research papers, original research notes, invited topical reviews (including Minireviews), and editorial commentary and news. The area of focus shall be concepts in basic pharmaceutical science and such topics as chemical processing of pharmaceuticals, including crystallization, lyophilization, chemical stability of drugs, pharmacokinetics, biopharmaceutics, pharmacodynamics, pro-drug developments, metabolic disposition of bioactive agents, dosage form design, protein-peptide chemistry and biotechnology specifically as these relate to pharmaceutical technology, and targeted drug delivery.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
