Kyle P. Flannery, Sylvia Safwat, Eli Matsell, Namarata Battula, Ahlam A. A. Hamed, Inaam N. Mohamed, Maha A. Elseed, Mahmoud Koko, Rayan Abubaker, Fatima Abozar, Liena E. O. Elsayed, Vikram Bhise, Robert S. Molday, Mustafa A. Salih, Ashraf Yahia, M. Chiara Manzini
{"title":"A novel missense variant in the ATPase domain of ATP8A2 and review of phenotypic variability of ATP8A2-related disorders caused by missense changes","authors":"Kyle P. Flannery, Sylvia Safwat, Eli Matsell, Namarata Battula, Ahlam A. A. Hamed, Inaam N. Mohamed, Maha A. Elseed, Mahmoud Koko, Rayan Abubaker, Fatima Abozar, Liena E. O. Elsayed, Vikram Bhise, Robert S. Molday, Mustafa A. Salih, Ashraf Yahia, M. Chiara Manzini","doi":"10.1007/s10048-024-00773-9","DOIUrl":null,"url":null,"abstract":"<p>ATPase, class 1, type 8 A, member 2 (ATP8A2) is a P4-ATPase with a critical role in phospholipid translocation across the plasma membrane. Pathogenic variants in <i>ATP8A2</i> are known to cause cerebellar ataxia, impaired intellectual development, and disequilibrium syndrome 4 (CAMRQ4) which is often associated with encephalopathy, global developmental delay, and severe motor deficits. Here, we present a family with two siblings born from a consanguineous, first-cousin union from Sudan presenting with global developmental delay, intellectual disability, spasticity, ataxia, nystagmus, and thin corpus callosum. Whole exome sequencing revealed a homozygous missense variant in the nucleotide binding domain of ATP8A2 (p.Leu538Pro) that results in near complete loss of protein expression. This is in line with other missense variants in the same domain leading to protein misfolding and loss of ATPase function. In addition, by performing diffusion-weighted imaging, we identified bilateral hyperintensities in the posterior limbs of the internal capsule suggesting possible microstructural changes in axon tracts that had not been appreciated before and could contribute to the sensorimotor deficits in these individuals.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"44 1","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2024-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-024-00773-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
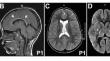
ATPase, class 1, type 8 A, member 2 (ATP8A2) is a P4-ATPase with a critical role in phospholipid translocation across the plasma membrane. Pathogenic variants in ATP8A2 are known to cause cerebellar ataxia, impaired intellectual development, and disequilibrium syndrome 4 (CAMRQ4) which is often associated with encephalopathy, global developmental delay, and severe motor deficits. Here, we present a family with two siblings born from a consanguineous, first-cousin union from Sudan presenting with global developmental delay, intellectual disability, spasticity, ataxia, nystagmus, and thin corpus callosum. Whole exome sequencing revealed a homozygous missense variant in the nucleotide binding domain of ATP8A2 (p.Leu538Pro) that results in near complete loss of protein expression. This is in line with other missense variants in the same domain leading to protein misfolding and loss of ATPase function. In addition, by performing diffusion-weighted imaging, we identified bilateral hyperintensities in the posterior limbs of the internal capsule suggesting possible microstructural changes in axon tracts that had not been appreciated before and could contribute to the sensorimotor deficits in these individuals.
ATPase, class 1, type 8 A, member 2 (ATP8A2)是一种P4-ATPase,在磷脂跨质膜转运中起着关键作用。已知 ATP8A2 的致病变异可导致小脑共济失调、智力发育受损和失衡综合征 4(CAMRQ4),而失衡综合征 4 通常与脑病、全面发育迟缓和严重运动障碍有关。在这里,我们介绍了一个由来自苏丹的嫡亲表兄弟姐妹所组成的家庭,该家庭中的两兄妹均表现为全面发育迟缓、智力障碍、痉挛、共济失调、眼球震颤和胼胝体薄弱。全外显子组测序发现,ATP8A2的核苷酸结合域存在一个同源错义变异(p.Leu538Pro),导致蛋白表达几乎完全丧失。这与同一结构域中导致蛋白质错误折叠和 ATPase 功能丧失的其他错义变异一致。此外,通过弥散加权成像,我们在内囊后肢发现了双侧高密度,这表明轴突束可能发生了微结构变化,而这种变化以前从未被发现过,并可能导致这些患者出现感觉运动障碍。
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
