Zhen Li, Subhamoy Bhowmik, Luca Sagresti, Giuseppe Brancato, Madelyn Smith, David E Benson, Pengfei Li, Kenneth M Merz
{"title":"Simulating Metal-Imidazole Complexes.","authors":"Zhen Li, Subhamoy Bhowmik, Luca Sagresti, Giuseppe Brancato, Madelyn Smith, David E Benson, Pengfei Li, Kenneth M Merz","doi":"10.1021/acs.jctc.4c00581","DOIUrl":null,"url":null,"abstract":"<p><p>One commonly observed binding motif in metalloproteins involves the interaction between a metal ion and histidine's imidazole side chains. Although previous imidazole-M(II) parameters established the flexibility and reliability of the 12-6-4 Lennard-Jones (LJ)-type nonbonded model by simply tuning the ligating atom's polarizability, they have not been applied to multiple-imidazole complexes. To fill this gap, we systematically simulate multiple-imidazole complexes (ranging from one to six) for five metal ions (Co(II), Cu(II), Mn(II), Ni(II), and Zn(II)) which commonly appear in metalloproteins. Using extensive (40 ns per PMF window) sampling to assemble free energy association profiles (using OPC water and standard HID imidazole charge models from AMBER) and comparing the equilibrium distances to DFT calculations, a new set of parameters was developed to focus on energetic and geometric features of multiple-imidazole complexes. The obtained free energy profiles agree with the experimental binding free energy and DFT calculated distances. To validate our model, we show that we can close the thermodynamic cycle for metal-imidazole complexes with up to six imidazole molecules in the first solvation shell. Given the success in closing the thermodynamic cycles, we then used the same extended sampling method for six other metal ions (Ag(I), Ca(II), Cd(II), Cu(I), Fe(II), and Mg(II)) to obtain new parameters. Since these new parameters can reproduce the one-imidazole geometry and energy accurately, we hypothesize that they will reasonably predict the binding free energy of higher-level coordination numbers. Hence, we did not extend the analysis of these ions up to six imidazole complexes. Overall, the results shed light on metal-protein interactions by emphasizing the importance of ligand-ligand interaction and metal-π-stacking within metalloproteins.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"6706-6716"},"PeriodicalIF":5.5000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11325557/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00581","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
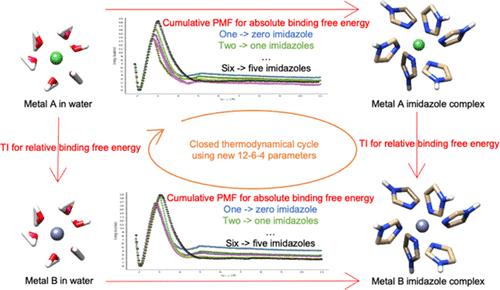
One commonly observed binding motif in metalloproteins involves the interaction between a metal ion and histidine's imidazole side chains. Although previous imidazole-M(II) parameters established the flexibility and reliability of the 12-6-4 Lennard-Jones (LJ)-type nonbonded model by simply tuning the ligating atom's polarizability, they have not been applied to multiple-imidazole complexes. To fill this gap, we systematically simulate multiple-imidazole complexes (ranging from one to six) for five metal ions (Co(II), Cu(II), Mn(II), Ni(II), and Zn(II)) which commonly appear in metalloproteins. Using extensive (40 ns per PMF window) sampling to assemble free energy association profiles (using OPC water and standard HID imidazole charge models from AMBER) and comparing the equilibrium distances to DFT calculations, a new set of parameters was developed to focus on energetic and geometric features of multiple-imidazole complexes. The obtained free energy profiles agree with the experimental binding free energy and DFT calculated distances. To validate our model, we show that we can close the thermodynamic cycle for metal-imidazole complexes with up to six imidazole molecules in the first solvation shell. Given the success in closing the thermodynamic cycles, we then used the same extended sampling method for six other metal ions (Ag(I), Ca(II), Cd(II), Cu(I), Fe(II), and Mg(II)) to obtain new parameters. Since these new parameters can reproduce the one-imidazole geometry and energy accurately, we hypothesize that they will reasonably predict the binding free energy of higher-level coordination numbers. Hence, we did not extend the analysis of these ions up to six imidazole complexes. Overall, the results shed light on metal-protein interactions by emphasizing the importance of ligand-ligand interaction and metal-π-stacking within metalloproteins.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
