Thomas Dalton Andress, David M Stanbury, David A Dixon
{"title":"Calculated Aqueous Reduction Potentials of Neutral and Anionic Halogen Diatomic Molecules.","authors":"Thomas Dalton Andress, David M Stanbury, David A Dixon","doi":"10.1021/acs.jpca.4c04037","DOIUrl":null,"url":null,"abstract":"<p><p>The free energy of hydration, aqueous, and gas phase electron affinity and aqueous reduction potentials of F<sub>2</sub>, Cl<sub>2</sub>, Br<sub>2</sub>, I<sub>2</sub>, ClF, BrF, IF, BrCl, ICl, IBr, and their corresponding anions were calculated using an electronic structure approach previously developed for these properties for X<sup>•</sup> and XO<sup>•</sup>, where X is a halogen which yielded excellent results. The gas phase electron affinities were calculated at the Feller-Peterson-Dixon level based on complete basis set extrapolation of CCSD(T) results with additional corrections. The agreement with the available experimental data is excellent, and the calculations provide a complete set of reliable electron affinities for these diatomic halogens. The hybrid solvation approach uses single point implicit solvation calculations on gas phase optimized clusters with explicit solvent molecules. The gas phase energy calculations were performed using MP2 and CCSD(T)-F12b for tetramer clusters (four explicit waters) and MP2 for octamer clusters (eight explicit waters). The final redox potentials were obtained at the MP2/aug-cc-pVTZ (aT) with a self-consistent reaction field (SMD) level using the octamer clusters. The aqueous reduction potentials of the neutral diatomic halogens are predicted within 0.06 V of the experiment for diatomic neutrals. The same agreement of 0.06 V is predicted for the redox potential resulting from dissociation electron attachment of the diatomic halogen anions. The current work extends reduction potentials for multiple redox couples for which no experimental data is available, for example, those containing iodine and the interhalogen anions. F<sub>2</sub><sup>•<b>-</b></sup> is predicted to dissociate for its lowest energy structure in both the tetramer and octamer clusters to form solvated F<b><sup>-</sup></b>, HF, and OH<sup>•</sup>.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":" ","pages":"6494-6509"},"PeriodicalIF":2.8000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c04037","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/31 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
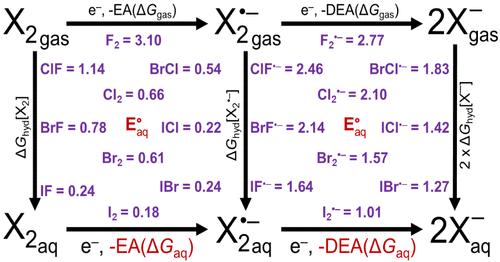
The free energy of hydration, aqueous, and gas phase electron affinity and aqueous reduction potentials of F2, Cl2, Br2, I2, ClF, BrF, IF, BrCl, ICl, IBr, and their corresponding anions were calculated using an electronic structure approach previously developed for these properties for X• and XO•, where X is a halogen which yielded excellent results. The gas phase electron affinities were calculated at the Feller-Peterson-Dixon level based on complete basis set extrapolation of CCSD(T) results with additional corrections. The agreement with the available experimental data is excellent, and the calculations provide a complete set of reliable electron affinities for these diatomic halogens. The hybrid solvation approach uses single point implicit solvation calculations on gas phase optimized clusters with explicit solvent molecules. The gas phase energy calculations were performed using MP2 and CCSD(T)-F12b for tetramer clusters (four explicit waters) and MP2 for octamer clusters (eight explicit waters). The final redox potentials were obtained at the MP2/aug-cc-pVTZ (aT) with a self-consistent reaction field (SMD) level using the octamer clusters. The aqueous reduction potentials of the neutral diatomic halogens are predicted within 0.06 V of the experiment for diatomic neutrals. The same agreement of 0.06 V is predicted for the redox potential resulting from dissociation electron attachment of the diatomic halogen anions. The current work extends reduction potentials for multiple redox couples for which no experimental data is available, for example, those containing iodine and the interhalogen anions. F2•- is predicted to dissociate for its lowest energy structure in both the tetramer and octamer clusters to form solvated F-, HF, and OH•.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
